WO2014196607A1 - 硬化性樹脂組成物、それを用いてなる構造接着剤、コーティング材又は繊維強化複合材料、それを発泡してなる発泡体、それを硬化してなる積層体、及びそれらの硬化物 - Google Patents
硬化性樹脂組成物、それを用いてなる構造接着剤、コーティング材又は繊維強化複合材料、それを発泡してなる発泡体、それを硬化してなる積層体、及びそれらの硬化物 Download PDFInfo
- Publication number
- WO2014196607A1 WO2014196607A1 PCT/JP2014/064998 JP2014064998W WO2014196607A1 WO 2014196607 A1 WO2014196607 A1 WO 2014196607A1 JP 2014064998 W JP2014064998 W JP 2014064998W WO 2014196607 A1 WO2014196607 A1 WO 2014196607A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- resin composition
- curable resin
- component
- mass
- parts
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09D—COATING COMPOSITIONS, e.g. PAINTS, VARNISHES OR LACQUERS; FILLING PASTES; CHEMICAL PAINT OR INK REMOVERS; INKS; CORRECTING FLUIDS; WOODSTAINS; PASTES OR SOLIDS FOR COLOURING OR PRINTING; USE OF MATERIALS THEREFOR
- C09D7/00—Features of coating compositions, not provided for in group C09D5/00; Processes for incorporating ingredients in coating compositions
- C09D7/40—Additives
- C09D7/65—Additives macromolecular
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G18/00—Polymeric products of isocyanates or isothiocyanates
- C08G18/06—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen
- C08G18/08—Processes
- C08G18/16—Catalysts
- C08G18/22—Catalysts containing metal compounds
- C08G18/24—Catalysts containing metal compounds of tin
- C08G18/244—Catalysts containing metal compounds of tin tin salts of carboxylic acids
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G18/00—Polymeric products of isocyanates or isothiocyanates
- C08G18/06—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen
- C08G18/28—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen characterised by the compounds used containing active hydrogen
- C08G18/40—High-molecular-weight compounds
- C08G18/42—Polycondensates having carboxylic or carbonic ester groups in the main chain
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G18/00—Polymeric products of isocyanates or isothiocyanates
- C08G18/06—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen
- C08G18/28—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen characterised by the compounds used containing active hydrogen
- C08G18/40—High-molecular-weight compounds
- C08G18/48—Polyethers
- C08G18/4825—Polyethers containing two hydroxy groups
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G18/00—Polymeric products of isocyanates or isothiocyanates
- C08G18/06—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen
- C08G18/28—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen characterised by the compounds used containing active hydrogen
- C08G18/40—High-molecular-weight compounds
- C08G18/48—Polyethers
- C08G18/4829—Polyethers containing at least three hydroxy groups
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G18/00—Polymeric products of isocyanates or isothiocyanates
- C08G18/06—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen
- C08G18/70—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen characterised by the isocyanates or isothiocyanates used
- C08G18/72—Polyisocyanates or polyisothiocyanates
- C08G18/74—Polyisocyanates or polyisothiocyanates cyclic
- C08G18/75—Polyisocyanates or polyisothiocyanates cyclic cycloaliphatic
- C08G18/751—Polyisocyanates or polyisothiocyanates cyclic cycloaliphatic containing only one cycloaliphatic ring
- C08G18/752—Polyisocyanates or polyisothiocyanates cyclic cycloaliphatic containing only one cycloaliphatic ring containing at least one isocyanate or isothiocyanate group linked to the cycloaliphatic ring by means of an aliphatic group
- C08G18/753—Polyisocyanates or polyisothiocyanates cyclic cycloaliphatic containing only one cycloaliphatic ring containing at least one isocyanate or isothiocyanate group linked to the cycloaliphatic ring by means of an aliphatic group containing one isocyanate or isothiocyanate group linked to the cycloaliphatic ring by means of an aliphatic group having a primary carbon atom next to the isocyanate or isothiocyanate group
- C08G18/755—Polyisocyanates or polyisothiocyanates cyclic cycloaliphatic containing only one cycloaliphatic ring containing at least one isocyanate or isothiocyanate group linked to the cycloaliphatic ring by means of an aliphatic group containing one isocyanate or isothiocyanate group linked to the cycloaliphatic ring by means of an aliphatic group having a primary carbon atom next to the isocyanate or isothiocyanate group and at least one isocyanate or isothiocyanate group linked to a secondary carbon atom of the cycloaliphatic ring, e.g. isophorone diisocyanate
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G18/00—Polymeric products of isocyanates or isothiocyanates
- C08G18/06—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen
- C08G18/70—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen characterised by the isocyanates or isothiocyanates used
- C08G18/72—Polyisocyanates or polyisothiocyanates
- C08G18/74—Polyisocyanates or polyisothiocyanates cyclic
- C08G18/76—Polyisocyanates or polyisothiocyanates cyclic aromatic
- C08G18/7657—Polyisocyanates or polyisothiocyanates cyclic aromatic containing two or more aromatic rings
- C08G18/7664—Polyisocyanates or polyisothiocyanates cyclic aromatic containing two or more aromatic rings containing alkylene polyphenyl groups
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J5/00—Manufacture of articles or shaped materials containing macromolecular substances
- C08J5/04—Reinforcing macromolecular compounds with loose or coherent fibrous material
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J5/00—Manufacture of articles or shaped materials containing macromolecular substances
- C08J5/24—Impregnating materials with prepolymers which can be polymerised in situ, e.g. manufacture of prepregs
- C08J5/241—Impregnating materials with prepolymers which can be polymerised in situ, e.g. manufacture of prepregs using inorganic fibres
- C08J5/243—Impregnating materials with prepolymers which can be polymerised in situ, e.g. manufacture of prepregs using inorganic fibres using carbon fibres
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J9/00—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof
- C08J9/0061—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof characterized by the use of several polymeric components
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J9/00—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof
- C08J9/02—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof using blowing gases generated by the reacting monomers or modifying agents during the preparation or modification of macromolecules
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L75/00—Compositions of polyureas or polyurethanes; Compositions of derivatives of such polymers
- C08L75/04—Polyurethanes
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09D—COATING COMPOSITIONS, e.g. PAINTS, VARNISHES OR LACQUERS; FILLING PASTES; CHEMICAL PAINT OR INK REMOVERS; INKS; CORRECTING FLUIDS; WOODSTAINS; PASTES OR SOLIDS FOR COLOURING OR PRINTING; USE OF MATERIALS THEREFOR
- C09D175/00—Coating compositions based on polyureas or polyurethanes; Coating compositions based on derivatives of such polymers
- C09D175/04—Polyurethanes
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09J—ADHESIVES; NON-MECHANICAL ASPECTS OF ADHESIVE PROCESSES IN GENERAL; ADHESIVE PROCESSES NOT PROVIDED FOR ELSEWHERE; USE OF MATERIALS AS ADHESIVES
- C09J11/00—Features of adhesives not provided for in group C09J9/00, e.g. additives
- C09J11/08—Macromolecular additives
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09J—ADHESIVES; NON-MECHANICAL ASPECTS OF ADHESIVE PROCESSES IN GENERAL; ADHESIVE PROCESSES NOT PROVIDED FOR ELSEWHERE; USE OF MATERIALS AS ADHESIVES
- C09J175/00—Adhesives based on polyureas or polyurethanes; Adhesives based on derivatives of such polymers
- C09J175/04—Polyurethanes
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G2110/00—Foam properties
- C08G2110/0008—Foam properties flexible
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G2110/00—Foam properties
- C08G2110/0083—Foam properties prepared using water as the sole blowing agent
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G2350/00—Acoustic or vibration damping material
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2201/00—Foams characterised by the foaming process
- C08J2201/02—Foams characterised by the foaming process characterised by mechanical pre- or post-treatments
- C08J2201/022—Foams characterised by the foaming process characterised by mechanical pre- or post-treatments premixing or pre-blending a part of the components of a foamable composition, e.g. premixing the polyol with the blowing agent, surfactant and catalyst and only adding the isocyanate at the time of foaming
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2205/00—Foams characterised by their properties
- C08J2205/10—Rigid foams
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2375/00—Characterised by the use of polyureas or polyurethanes; Derivatives of such polymers
- C08J2375/04—Polyurethanes
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2409/00—Characterised by the use of homopolymers or copolymers of conjugated diene hydrocarbons
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2413/00—Characterised by the use of rubbers containing carboxyl groups
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2419/00—Characterised by the use of rubbers not provided for in groups C08J2407/00 - C08J2417/00
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2425/00—Characterised by the use of homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by an aromatic carbocyclic ring; Derivatives of such polymers
- C08J2425/02—Homopolymers or copolymers of hydrocarbons
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2433/00—Characterised by the use of homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical, or of salts, anhydrides, esters, amides, imides, or nitriles thereof; Derivatives of such polymers
- C08J2433/04—Characterised by the use of homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical, or of salts, anhydrides, esters, amides, imides, or nitriles thereof; Derivatives of such polymers esters
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2433/00—Characterised by the use of homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical, or of salts, anhydrides, esters, amides, imides, or nitriles thereof; Derivatives of such polymers
- C08J2433/18—Homopolymers or copolymers of nitriles
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2451/00—Characterised by the use of graft polymers in which the grafted component is obtained by reactions only involving carbon-to-carbon unsaturated bonds; Derivatives of such polymers
- C08J2451/04—Characterised by the use of graft polymers in which the grafted component is obtained by reactions only involving carbon-to-carbon unsaturated bonds; Derivatives of such polymers grafted on to rubbers
Definitions
- the present invention relates to a polyurethane-based curable resin composition having excellent adhesion and toughness without reducing rigidity, and in particular, a structural adhesive, a coating agent or a coating agent using the curable resin composition
- the present invention relates to a fiber reinforced composite material, a foam formed by foaming the curable resin composition, a laminate formed by curing the curable resin composition, and a cured product thereof.
- Adhesives based on polyurethane show good adhesion to a wide variety of adherends such as plastic, wood, and metal, and are a combination of various isocyanate group-containing compounds and various active hydrogen group-containing compounds. Can provide a wide range of mechanical properties from hard to soft, and is used in many applications.
- structural adhesives used in the assembly of automobiles and other vehicles and aircraft need to have high rigidity to enhance the torsional rigidity of the body as well as adhesion. Furthermore, the structural adhesive is required to be excellent in fracture toughness, which is an index for suppressing the adhesive fracture received during the collision.
- Patent Document 1 discloses a urethane resin adhesive suitable for structural adhesion of a vehicle. However, it is difficult for an adhesive mainly composed of polyurethane to achieve both high rigidity and adhesiveness.
- a highly hard coating agent is desired while improving brittleness, which is a defect of an epoxy-based coating material.
- urethane composite materials reinforced with reinforcing fibers are also required to have higher elastic modulus, and it is important to achieve both high elastic modulus and toughness.
- curable resin composition containing polyurethane as a main component it has been difficult for a curable resin composition containing polyurethane as a main component to achieve both high rigidity (for example, high elastic modulus) and adhesion and toughness.
- This invention is made
- the objective of this invention is providing the curable resin composition excellent in adhesiveness and toughness, without reducing the rigidity of the hardened
- Another object of the present invention is to provide a technique using the curable resin composition.
- the gist of the present invention is as follows.
- a curable resin composition comprising a polyol (A) having an average hydroxyl value of 200 to 1500 mg KOH / g, a polyisocyanate (B), and polymer fine particles (C).
- the curable resin composition according to [1] which contains a polyether polyol (a1) as the polyol (A).
- the polyisocyanate (B) is contained in an amount of 2 to 5000 parts by mass with respect to 100 parts by mass of the polyol (A). Any one of [1] to [7], wherein the polymer fine particles (C) are contained in an amount of 1 to 100 parts by mass with respect to 100 parts by mass of the total amount of the polyol (A) and the polyisocyanate (B).
- the component (C) has one or more core layers selected from the group consisting of diene rubbers, (meth) acrylate rubbers, and organosiloxane rubbers.
- composition The curable resin according to any one of [1] to [19], wherein the component (B) is a polyisocyanate having an average of 2.1 or more isocyanate groups per molecule.
- Composition [21] The curable resin composition according to [18], wherein the component (B) is at least one selected from the group consisting of alicyclic polyisocyanates and aliphatic polyisocyanates.
- component (B) is an alicyclic polyisocyanate.
- the composition further comprises 0.001 to 20 parts by mass of a curing catalyst (D) with respect to 100 parts by mass of the total amount of the components (A) and (B).
- the curable resin composition in any one of. [24] The curable resin composition according to [23], wherein the component (D) is an organotin compound. [25]
- the dehydrating agent (E) is further contained in an amount of 0.1 to 30 parts by mass with respect to 100 parts by mass of the total amount of the component (A) and the component (B) [1] to [24] ]
- the curable resin composition in any one of. [26] The ratio value ( ⁇ / ⁇ ) of the total molar amount ( ⁇ ) of the isocyanate groups in the component (B) to the total molar amount ( ⁇ ) of the hydroxyl groups in the component (A) is 0.7-1
- the reaction is performed when the equivalent ratio (NCO / active hydrogen-containing group) of the isocyanate (NCO) group of the component (B) to the active hydrogen-containing group of the component (A) is in the range of 1.05 to 5.0.
- the curable resin composition according to [27] which contains a urethane prepolymer having an isocyanate obtained as described above.
- a one-component moisture curable resin composition comprising the curable resin composition according to [28].
- the cured product according to [31] which has a glass transition temperature of 75 ° C. or higher.
- the curable resin composition of the present invention is characterized by containing a polyol (A), a polyisocyanate (B), and polymer fine particles (C), and preferably any one of the first aspect and the second aspect.
- the first aspect is a curable resin composition (I) containing a polyol (A) having an average hydroxyl value of 200 to 1500 mgKOH / g, a polyisocyanate (B), and polymer fine particles (C).
- the second embodiment contains a polyol (A), a polyisocyanate (B), and polymer fine particles (C).
- the polyester polyol must be contained, and the amount of the polyester polyol (a2) is
- the curable resin composition (II) is characterized by being 20 parts by mass or more of 100 parts by mass of the total amount of the polyol (A) component.
- a curing catalyst (D) may further be contained.
- the first aspect is particularly advantageous in that it exhibits higher rigidity (elastic modulus) and toughness than the conventional one, and the second aspect is particularly advantageous in that it exhibits an equal or higher adhesion than the conventional one. .
- the polyol (A) is used.
- a polyol (A) may be used independently and may be used together 2 or more types.
- the component (A) is a compound having two or more active hydrogens at the terminal, and is a bifunctional or higher functional polyol having a molecular weight of about 50 to 20,000, including aliphatic alcohols, aromatic alcohols, polyether polyols, Examples include polyester polyols, polyolefin polyols, and acrylic polyols.
- the average hydroxyl value of the polyol (A) component is 200 to 1500 mgKOH / g.
- polyether polyol (a1) as said polyol (A).
- Polyether polyol has low viscosity and excellent workability, and is excellent in the balance between hardness and toughness of the resulting cured product.
- the amount of the polyether polyol (a1) is preferably 50 parts by mass or more, more preferably 70 parts by mass or more, out of 100 parts by mass of the total amount of the polyol (A) component. More preferably, it is more preferably 90 parts by mass or more.
- the upper limit of the amount of the polyether polyol (a1) is, for example, 100 parts by mass.
- polyether polyol (a1) examples include, for example, ring-opening polymerization of ethylene oxide, propylene oxide, butylene oxide, styrene oxide, etc. in the presence of an initiator containing one or more of the following active hydrogens: And a random or block copolymer obtained by the above, and a mixture thereof.
- the initiator containing active hydrogen include ethylene glycol, diethylene glycol, propylene glycol, dipropylene glycol, 1,3-butanediol, 1,4-butanediol, 1,6-hexanediol, and neopentyl glycol.
- Diols such as bisphenol A; triols such as trimethylolethane, trimethylolpropane and glycerine; saccharides such as monosaccharides, oligosaccharides and polysaccharides; sorbitol; amines such as ammonia, ethylenediamine, urea, monomethyldiethanolamine and monoethyldiethanolamine And the like.
- the polyester polyol (a2) is necessarily contained as the polyol (A) component, and the amount of the polyester polyol (a2) is (A) It is essential that the total amount of the components is 20 parts by mass or more in 100 parts by mass.
- the amount of (a2) is more preferably 25 parts by mass or more, more preferably 30 parts by mass or more, and particularly preferably 33 parts by mass or more, out of 100 parts by mass of the total amount of component (A).
- the amount of (a2) is, for example, 100 parts by mass or less, preferably 90 parts by mass or less, more preferably 80 parts by mass or less, and still more preferably 70 parts by mass or less.
- the resulting cured product may have a low elastic modulus.
- Polyester polyol is excellent in adhesiveness and excellent in the balance between hardness and toughness of the resulting cured product.
- the average hydroxyl value of the component (A) in the curable resin composition (II) is preferably 20 to 1000 mgKOH / g.
- the average hydroxyl value is more preferably 50 mgKOH / g or more, further preferably 100 mgKOH / g or more, still more preferably 130 mgKOH / g or more, more preferably 500 mgKOH / g or less, still more preferably 300 mgKOH / g or less, More preferably, it is 200 mgKOH / g or less. If the average hydroxyl value is less than 20 mg KOH / g, the resulting cured product may have a low modulus of elasticity, or the effect of improving toughness and elongation due to the component (C) may be small. If the average hydroxyl value is greater than 1000 mg KOH / g, The toughness may be low.
- the functional group of the polyester polyol and the polyether polyol is preferably a bifunctional or higher functional group.
- the hydroxyl value can be obtained by a measuring method based on the standard of JIS K1557-1.
- polyester polyol (a2) include, for example, polybasic acids such as maleic acid, fumaric acid, adipic acid, sebacic acid, phthalic acid, dodecanedioic acid, isophthalic acid, azelaic acid, and acid anhydrides thereof, and ethylene glycol.
- Esterification catalyst with polyhydric alcohols such as propylene glycol, 1,4-butanediol, 1,6-hexanediol, diethylene glycol, dipropylene glycol, neopentyl glycol, 3-methyl-1,5-pentanediol
- a polymer obtained by polycondensation in the temperature range of 150 to 270 ° C. in the presence of include ring-opening polymers such as ⁇ -caprolactone and valerolactone, and active hydrogen compounds having two or more active hydrogens such as polycarbonate diol and castor oil.
- the polyhydric alcohol can be used alone or in combination of two or more selected from saccharides, aliphatic alcohols, aromatic alcohols, polyolefin-type polyols, acrylic polyols and the like.
- saccharide examples include saccharides such as monosaccharides, oligosaccharides, and polysaccharides.
- the aliphatic alcohol may be a dihydric alcohol or a trihydric or higher alcohol (trihydric alcohol, tetrahydric alcohol, etc.).
- dihydric alcohol include ethylene glycol, propylene glycol, and 1,3-propanediol.
- Alkylene glycols such as 1,3-butanediol, 1,4-butanediol, 1,5-pentanediol, 1,6-hexanediol, 3-methyl-1,5-pentanediol, neopentyl glycol, etc.
- the trihydric alcohol include glycerin, trimethylolpropane, trimethylolethane, 1,2,6-hexanetriol (particularly, a trihydric alcohol having about 3 to 10 carbon atoms).
- Examples of the tetrahydric alcohol include pentaerythritol and diglycerin.
- aromatic alcohol examples include bisphenols such as bisphenol A and bisphenol F; biphenyls such as dihydroxybiphenyl; polyhydric phenols such as hydroquinone and phenol formaldehyde condensate; and naphthalenediol.
- polystyrene-type polyol examples include polybutadiene polyol, polyisoprene polyol and hydrogenated products thereof.
- acrylic polyol examples include a monomer having a hydroxyl group such as hydroxyethyl (meth) acrylate, hydroxybutyl (meth) acrylate, and vinylphenol, and a general-purpose monomer such as n-butyl (meth) acrylate and 2-ethylhexyl (meth) acrylate. And a copolymer thereof, and a mixture thereof.
- a trifunctional or higher polyfunctional polyol having three or more hydroxyl groups in one molecule is preferable because it has high reactivity in curing and the resulting cured product has high hardness.
- the component preferably contains a trifunctional or higher polyfunctional polyol.
- the content of the trifunctional or higher polyfunctional polyol is preferably 20 parts by mass or more, more preferably 30 parts by mass or more, and more preferably 50 parts by mass or more out of the total amount (total 100 parts by mass) of the component (A). More preferred is 70 parts by mass or more.
- the content of the trifunctional or higher polyfunctional polyol is, for example, 100 parts by mass or less, preferably 90 parts by mass or less, and more preferably 80 parts by mass or less. If it is less than 20 mass parts, the rigidity of the hardened
- the content of the component (A) as a constituent component in the curable resin composition of the present invention is preferably 10% by mass or more, more preferably 15% by mass or more, and more preferably 20% by mass of the total amount of the curable resin composition.
- the above is more preferable, and 25% by mass or more is particularly preferable.
- the content is preferably 90% by mass or less of the total amount of the curable resin composition, more preferably 85% by mass or less, still more preferably 80% by mass or less, and particularly preferably 75% by mass or less. If it is less than 10 mass% or more than 90 mass%, the toughness and adhesiveness of the resulting cured product may be lowered.
- 25 mass% or more is preferable with respect to the total mass except an inorganic component from this curable resin composition, 25 mass% or more is more preferable, 30 mass% or more is more preferable, and 35 mass% or more is still more.
- 40 mass% or more is particularly preferable.
- the content of the component (A) is preferably 75% by mass or less, more preferably 70% by mass or less, and still more preferably 65% by mass or less, based on the total mass excluding the inorganic component from the curable resin composition. Especially preferably, it is 60 mass% or less. If it is less than 25% by mass or more than 75% by mass, the toughness and adhesion of the resulting cured product may deteriorate.
- polyisocyanate (B) In the curable resin composition of the present invention, polyisocyanate (B) is used. Polyisocyanate (B) may be used independently and may be used together 2 or more types. The polyisocyanate (B) is an essential component that reacts with the component (A) of the present invention to form polyurethane in the curable resin composition.
- the number of isocyanate groups in the component (B) is 2 or more per molecule, but the component (B) is a polyisocyanate compound having an average of 2.1 or more isocyanate groups per molecule.
- the combination with the later-described component (C) is preferable because the effect of improving the physical properties (adhesiveness, toughness, impact resistance) of the resulting cured product is remarkable.
- the average number of isocyanate groups is more preferably 2.3 or more per molecule, still more preferably 2.5 or more, more preferably 20 or less, and even more preferably 10 It is as follows.
- Diisocyanate compounds 1,3,5-triisocyanate cyclohexane, 1,3,5-trimethyl isocyanate cyclohexane, 3-isocyanate Tyl-3,3,5-trimethylcyclohexyl isocyanate, 2- (3-isocyanatopropyl) -2,5-di (isocyanatemethyl) -bicyclo [2,2,1] heptane, 2- (3-isocyanatopropyl)- 2,6-di (isocyanatomethyl) -bicyclo [2,2,1] heptane, 3- (3-isocyanatopropyl) -2,5-di (isocyanatomethyl) -bicyclo [2,2,1] heptane, 5 -(2-isocyanatoethyl) -2-isocyanatomethyl-3- (3-isocyanatopropyl) -bicyclo [2,2,1] heptane, 6- (2-isocyanato
- aliphatic, alicyclic, or araliphatic polyisocyanates When yellowing becomes a problem when using these polyisocyanate compounds, it is preferable to use aliphatic, alicyclic, or araliphatic polyisocyanates, and aliphatic polyisocyanates or alicyclic polyisocyanates are used. More preferred.
- an aliphatic polyisocyanate is used as the component (B) and an organotin compound in the component (D) described later is used as the urethane curing catalyst component, there is no yellowing and excellent weather resistance and curability. Is particularly preferable.
- the (A) component and the (B) component can be reacted for the first time at the time of curing the curable resin composition, but a part or all of the (A) component and the (B) component are reacted in advance.
- the so-called prepolymer can be used in a curable resin composition.
- the component (C) may be dispersed in the component (A) and then reacted with the component (B) to be prepolymerized.
- the said curable resin composition may contain the urethane prepolymer obtained by reaction of the said (A) component and the said (B) component.
- the curable resin composition that consumed all the active hydrogen-containing groups of the component (A) in the curable resin composition is cured by the reaction between the isocyanate groups in the prepolymer and moisture in the air. It can be used as a one-component moisture-curable resin composition that can be used. That is, the present invention includes a one-component moisture curable resin composition comprising the curable resin composition.
- the polymer fine particles preferably do not contain powdered polymer fine particles or coagulated / dried polymer fine particles in the curable resin composition or the cured product thereof, and are derived from an aqueous latex as described later. Further, it is preferably composed of polymer fine particles that are not coagulated and dried. With such polymer fine particles, the dispersed state of primary particles can be easily realized, while with the fine polymer particles of powder, it is difficult to realize the dispersed state of primary particles.
- Methacrylates methacrylic monomers containing methacrylic acid derivatives such as methacrylonitrile; certain acrylic esters such as isobornyl acrylate and tert-butyl acrylate; acrylic monomers containing acrylic acid derivatives such as acrylonitrile.
- Further examples include monomers having a Tg of 120 ° C. or higher, such as acrylamide, isopropylacrylamide, N-vinylpyrrolidone, dicyclopentanyl methacrylate, 2-methyl-2-adamantyl methacrylate, 1-adamantyl acrylate, and 1-adamantyl methacrylate.
- a group, an oxetane group, a hydroxyl group an amino group, an imide group, a carboxylic acid group, a carboxylic anhydride group, a cyclic ester group, a cyclic amide group, a benzoxazine group, and a cyanate ester group
- the graft ratio of the shell layer is preferably 70% or more (more preferably 80% or more, and further 90% or more). When the graft ratio is less than 70%, the viscosity of the liquid resin composition may increase.
- the method for calculating the graft ratio is as follows.
- an aqueous latex containing polymer fine particles is coagulated and dehydrated, and finally dried to obtain polymer fine particle powder.
- 2 g of the polymer fine particle powder was immersed in 100 g of methyl ethyl ketone (MEK) at 23 ° C. for 24 hours, and then the MEK soluble component was separated from the MEK insoluble component, and the methanol insoluble component was further separated from the MEK soluble component. And it calculated by calculating
- MEK methyl ethyl ketone
- alkyl or aryl sulfonic acid represented by dioctylsulfosuccinic acid and dodecylbenzenesulfonic acid
- alkyl or arylether sulfonic acid alkyl or aryl represented by dodecylsulfuric acid, and the like.
- a curing catalyst can be used as the component (D) as necessary.
- the curing catalyst is not particularly limited and includes a catalyst that promotes a commonly used urethanization reaction. Specific examples include tin 2-ethylhexanoate, tin versatate, bismuth 2-ethylhexanoate, potassium acetate, potassium octylate, lead octylate, lead naphthenate, nickel naphthenate, cobalt octylate and the like.
- the said (D) component is an organotin compound from a viewpoint of yellowing suppression.
- a salt of the amine compound such as DBU octylate and an organic acid such as carboxylic acid or phenol is effective as a latent curable catalyst.
- the amount of the curing catalyst is preferably 0.001 to 20 parts by mass, more preferably 0.01 to 5 parts by mass, and still more preferably, with respect to 100 parts by mass of the total amount of the component (A) and the component (B). 0.05 to 2 parts by mass, particularly preferably 0.1 to 1 part by mass. If it is less than 0.001 part by mass, curing may be delayed. If it exceeds 20 parts by mass, curing may be too early and handling may be difficult.
- a filler can be added to the curable resin composition of the present invention.
- Fillers include reinforcing silica such as fumed silica, precipitated silica, crystalline silica, fused silica, dolomite, anhydrous silicic acid, hydrous silicic acid, and carbon black; heavy calcium carbonate, colloidal calcium carbonate, carbonic acid Magnesium, barium carbonate, barium sulfate, diatomaceous earth, calcined clay, clay, talc, barite, anhydrous gypsum, titanium oxide, bentonite, organic bentonite, ferric oxide, aluminum fine powder, flint powder, zinc oxide, activated zinc white, Fillers such as mica, zinc white, lead white, lithopone, zinc sulfide, shirasu balloon, glass microballoon, organic microballoon of phenol resin and vinylidene chloride resin, PVC powder, PMMA powder; asbestos, glass fiber and filament And the like,
- coloring pigments such as titanium oxide, lead chromate, chromium oxide, ultramarine, cobalt blue, cyanine blue, cyanine green, lake red, and quinacridone red can also be used.
- the amount used is preferably 1 to 250 parts by weight, more preferably 10 to 200 parts by weight, based on 100 parts by weight of the total amount of components (A) and (B).
- the amount used is preferably 5 to 200 parts by weight, more preferably 10 to 100 parts by weight with respect to 100 parts by weight as the total of the components (A) and (B). Used in.
- ANTI-TERRA registered trademark
- DISPERBYK registered trademark
- DISPERBYK registered trademark
- DISPERBYK registered trademark
- DISPERBYK registered trademark
- DISPERBYK registered trademark
- DISPERBYK registered trademark
- DISPERBYK registered trademark
- DISPERBYK registered trademark
- DISPERBYK registered trademark
- DISPERBYK registered trademark
- DISPERBYK registered trademark
- DISPARLON registered trademark
- DISPARLON registered trademark
- DISPARLON registered trademark
- DISPARLON registered trademark
- DISPARLON registered trademark
- DISPARLON registered trademark
- DISPARLON registered trademark
- DISPARLON registered trademark
- DISPARLON registered trademark
- DISPARLON registered trademark
- DISPARLON registered trademark
- DISPARLON registered trademark
- DISPARLON registered trademark
- DISPARLON registered trademark
- DISPARLON registered trademark
- DISPARLON registered trademark
- DISPARLON registered trademark
- DISPARLON registered trademark
- DISPARLON registered trademark
- DISPARLON registered trademark
- DISPARLON registered trademark
- DISPARLON registered trademark
- DISPARLON registered trademark
- DISPARLON registered trademark
- DISPARLON registered trademark
- DISPARLON registered trademark
- DISPARLON registered trademark
- DISPARLON registered trademark
- DISPARLON registered trademark
- DISPARLON registered trademark
- DISPARLON registered trademark
- DISPARLON registered trademark
- the number average molecular weight of the dispersant is preferably 1000 to 100,000. If it is less than 1000, sufficient dispersion stability may not be obtained, and if it exceeds 100,000, the viscosity may be too high and handling may be difficult. More preferably, it is 2000 to 50,000, and still more preferably 4000 to 50,000.
- the amount used is preferably 0.1 to 10 parts by mass, more preferably 0.2 to 3 parts per 100 parts by mass of the total amount of component (A) and component (B). It is used in parts by weight, more preferably in the range of 0.3 to 1 part by weight.
- BYK (registered trademark) -1752 BYK (registered trademark) -1790
- BYK (registered trademark) -060N BYK (registered trademark) -063, BYK (registered trademark) -065, BYK (registered trademark).
- a plasticizer can be added to the curable resin composition of the present invention as necessary. By adding a plasticizer, the viscosity and slump property of the curable resin composition and the mechanical properties such as tensile strength and elongation of the cured product obtained by curing the composition can be adjusted.
- the amount of the plasticizer used is the components (A) and (B). 100 mass parts or less are preferable with respect to 100 mass parts of total components, 50 mass parts or less are more preferable, 30 mass parts or less are still more preferable, 10 mass parts or less are especially preferable, It is most preferable not to contain.
- a solvent can be used as necessary for the purpose of reducing the viscosity of the composition, increasing thixotropy, and improving workability.
- the solvent is not particularly limited, and various compounds can be used. Specific examples include hydrocarbon solvents such as toluene, xylene, heptane, hexane, petroleum solvents, halogen solvents such as trichloroethylene, ester solvents such as ethyl acetate and butyl acetate, acetone, methyl ethyl ketone, and methyl isobutyl ketone.
- the blending amount of the solvent is preferably 50 parts by mass or less, more preferably 30 parts by mass or less, with respect to 100 parts by mass of the total amount of the components (A) and (B). It is more preferable that the amount is not more than part, and it is particularly preferable that no solvent is contained.
- the component (A) and the component (B) of the present invention can select a material having a relatively low viscosity, they are non-aqueous and / or non-solvent type (or high solid type having a low solvent content).
- the aqueous emulsion composition requires a long time for film formation at low temperatures or high humidity, and is difficult to apply at cold temperatures, so the curable composition of the present invention is a non-aqueous curable composition. Is preferred.
- the curable composition of the present invention is preferably a non-solvent (or high solid) curable composition.
- the content of water in the curable composition of the present invention is preferably 10 parts by mass or less with respect to 100 parts by mass of the total amount of the component (A) and the component (B). 1 mass part or less is more preferable, 0.1 mass part or less is still more preferable, and it is the most preferable not to contain water substantially. If the water content exceeds this range, the physical properties of the cured product may deteriorate.
- a tackifier can be added to the curable resin composition of the present invention.
- a tackifier can be added to the curable resin composition of the present invention.
- a tackifier What is normally used regardless of solid and liquid at normal temperature can be used.
- Specific examples include styrene block copolymers, hydrogenated products thereof, phenol resins, modified phenol resins (for example, cashew oil modified phenol resin, tall oil modified phenol resin, etc.), terpene phenol resins, xylene-phenol resins, cyclohexane Pentadiene-phenol resin, coumarone indene resin, rosin resin, rosin ester resin, hydrogenated rosin ester resin, xylene resin, low molecular weight polystyrene resin, styrene copolymer resin, petroleum resin (for example, C5 hydrocarbon resin, C9) Hydrocarbon resin, C5C9 hydrocarbon copolymer resin, etc.), hydrogenated petroleum resin,
- Styrene block copolymers and their hydrogenated products include styrene-butadiene-styrene block copolymers (SBS), styrene-isoprene-styrene block copolymers (SIS), and styrene-ethylenebutylene-styrene block copolymers.
- SBS styrene-butadiene-styrene block copolymers
- SIS styrene-isoprene-styrene block copolymers
- SEBS styrene-ethylenebutylene-styrene block copolymer
- SEPS styrene-ethylenepropylene-styrene block copolymer
- SIBS styrene-isobutylene-styrene block copolymer
- the above tackifiers may be used alone or in combination of two or more.
- the amount used is preferably 5 to 100 parts by weight, more preferably 10 to 50 parts by weight, based on 100 parts by weight of the total amount of components (A) and (B). Used in the range of
- a leveling agent can be added to the composition of this invention as needed.
- Commercially available leveling agents can be used.
- Commercially available products include, for example, BYKETOL (registered trademark) -OK, BYKETOL (registered trademark) -SPECIAL, BYKETOL (registered trademark) -AQ, BYKETOL (registered trademark) -WS (all manufactured by BYK Chemie), DISPARLON (registered trademark) 1970, DISPARLON (registered trademark) 230, DISPARLON (registered trademark) LF-1980, DISPARLON (registered trademark) LF-1982, DISPARLON (registered trademark) LF-1983, DISPARLON (registered trademark) LF-1984, DISPARLON (registered trademark) LF-1985 (both manufactured by Enomoto Kasei Co., Ltd.).
- the amount used is preferably 0.05 to 10 parts by mass, more preferably 0.2 to 5 parts per 100 parts by mass of the total amount of the component (A) and the component (B). It is used in parts by weight, more preferably in the range of 0.3 to 3 parts by weight.
- a thixotropic agent may be added to the curable resin composition of the present invention as necessary to prevent sagging and improve workability.
- the anti-sagging agent is not particularly limited, and examples thereof include polyamide waxes; hydrogenated castor oil derivatives; metal soaps such as calcium stearate, aluminum stearate, and barium stearate.
- the fumed silica shown as a filler can also be used as a thixotropic agent.
- rubber powder having a particle diameter of 10 to 500 ⁇ m as described in JP-A-11-349916 or organic fiber as described in JP-A-2003-155389 is used, thixotropy is high. A composition having good workability can be obtained.
- These thixotropic agents may be used alone or in combination of two or more.
- the amount used is, for example, in the range of 0.1 to 20 parts by mass with respect to 100 parts by mass of the total amount of components (A) and (B).
- epoxy resin examples include bisphenol A type epoxy resin, bisphenol F type epoxy resin, novolac type epoxy resin, glycidyl ester type epoxy resin, hydrogenated bisphenol A (or F) type epoxy resin, glycidyl ether type epoxy resin, amino-containing Known epoxy resins such as epoxy compounds obtained by addition reaction of bisphenol A (or F), polybasic acids and the like to glycidyl ether resins and these epoxy resins can be mentioned.
- the amount used is, for example, in the range of 0.1 to 30 parts by mass with respect to 100 parts by mass of the total amount of the components (A) and (B).
- an antioxidant antioxidant
- cured material can be improved.
- antioxidant antioxidant
- examples of the antioxidant include hindered phenols, monophenols, bisphenols, and polyphenols, with hindered phenols being particularly preferred.
- the amount used is preferably in the range of 0.1 to 10 parts by mass, more preferably 0.1 to 10 parts by mass relative to 100 parts by mass of the total amount of the components (A) and (B). 2 to 5 parts by mass.
- a light stabilizer can be used as necessary.
- Use of a light stabilizer can prevent photooxidation degradation of the cured product.
- the light stabilizer include benzotriazole-based, hindered amine-based, and benzoate-based compounds, with hindered amine-based compounds being more preferable.
- a tertiary amine-containing hindered amine light stabilizer is preferably used for improving the storage stability of the composition.
- Tinuvin (registered trademark) 622LD Tinuvin (registered trademark) 144, CHIMASSORB (registered trademark) 119FL (all manufactured by BASF); MARK LA-57, LA-62, LA- 67, LA-63 (all of which are manufactured by ADEKA CORPORATION); SANOR (registered trademark) LS-765, LS-292, LS-2626, LS-1114, LS-744 (all of which are manufactured by Sankyo Corporation), etc.
- a light stabilizer can be illustrated.
- the amount used is preferably in the range of 0.1 to 10 parts by mass, more preferably 0.1 to 10 parts by mass relative to 100 parts by mass of the total amount of the components (A) and (B). 2 to 5 parts by mass.
- An ultraviolet absorber can be used in the curable resin composition of the present invention as necessary. When the ultraviolet absorber is used, the surface weather resistance of the cured product can be enhanced. Examples of ultraviolet absorbers include benzophenone-based, benzotriazole-based, salicylate-based, substituted tolyl-based, and metal chelate-based compounds, with benzotriazole-based compounds being particularly preferable.
- the ultraviolet absorber When used, it is preferably used in the range of 0.1 to 10 parts by mass, more preferably 0.1 to 10 parts by mass with respect to 100 parts by mass of the total amount of the components (A) and (B). 2 to 5 parts by mass. It is preferable to use a phenolic or hindered phenolic antioxidant, a hindered amine light stabilizer and a benzotriazole ultraviolet absorber in combination.
- a silane coupling agent can be added to the curable resin composition of the present invention. Adhesiveness can be improved by adding a silane coupling agent. Specific examples include ⁇ -isocyanatopropyltrimethoxysilane, ⁇ -isocyanatopropyltriethoxysilane, ⁇ -isocyanatopropylmethyldiethoxysilane, ⁇ -isocyanatopropylmethyldimethoxysilane, (isocyanatemethyl) trimethoxysilane, and (isocyanatemethyl).
- Isocyanate group-containing silanes such as dimethoxymethylsilane, (isocyanatemethyl) triethoxysilane, (isocyanatemethyl) diethoxymethylsilane; ⁇ -aminopropyltrimethoxysilane, ⁇ -aminopropyltriethoxysilane, ⁇ -aminopropyltriiso Propoxysilane, ⁇ -aminopropylmethyldimethoxysilane, ⁇ -aminopropylmethyldiethoxysilane, ⁇ - (2-aminoethyl) aminopropyltrime Xysilane, ⁇ - (2-aminoethyl) aminopropylmethyldimethoxysilane, ⁇ - (2-aminoethyl) aminopropyltriethoxysilane, ⁇ - (2-aminoethyl) aminopropylmethyldiethoxysilane, ⁇ - (2- Aminoethyl)
- silanes such as N- (1,3-dimethylbutylidene) -3- (triethoxysilyl) -1-propanamine; ⁇ -mercaptopropyltrimethoxysilane, ⁇ -mercaptopropyltri Mercapto group-containing silanes such as ethoxysilane, ⁇ -mercaptopropylmethyldimethoxysilane, ⁇ -mercaptopropylmethyldiethoxysilane, mercaptomethyltrimethoxysilane, mercaptomethyltriethoxysilane; ⁇ -glycidoxypropyltrimethoxysilane, ⁇ -glycidoxypropyltriethoxysilane, ⁇ -glycidoxypropylmethyldimethoxysilane, ⁇ - (3,4-epoxycyclohexyl) ethyltrimethoxysilane, ⁇ - (3,4-epoxy
- halogen-containing silanes such as chloropropyl trimethoxy silane.
- amino-modified silyl polymers silylated amino polymers, unsaturated aminosilane complexes, phenylamino long-chain alkylsilanes, aminosilylated silicones, silylated polyesters, and the like, which are derivatives of these, can also be used as silane coupling agents.
- the reaction product of the silane coupling agent include a reaction product of aminosilane and epoxysilane, a reaction product of aminosilane and isocyanate silane, and a partial condensate of various silane coupling agents.
- the amount used is preferably about 0.1 to 15 parts by weight, and about 0.5 to 10 parts by weight with respect to 100 parts by weight as the total of component (A) and component (B). Is more preferable, and about 1 to 5 parts by mass is particularly preferable. If the blending amount is below this range, the adhesion and storage stability may not be sufficient. On the other hand, if the blending amount exceeds this range, the strength of the cured product may decrease.
- a dehydrating agent can be added to the curable resin composition of the present invention as the component (E) as necessary. By adding a dehydrating agent, moisture present in the composition can be removed, and storage stability and foaming during curing are improved.
- a dehydrating agent include vinyltrimethoxysilane, calcium oxide, zeolite, p-toluenesulfonyl isocyanate, oxazolidines such as 3-ethyl-2-methyl-2- (3-methylbutyl) -1,3-oxazolidine, and the like. These can be used alone or in combination of two or more.
- ⁇ Other ingredients> In this invention, another compounding component can be used as needed.
- other blending components include hydrolysis stabilizers, titanate coupling agents, aluminate coupling agents, mold release agents, antistatic agents, lubricants, low shrinkage agents, silicone surfactants, and the like.
- the curable resin composition of the present invention is a composition containing polymer fine particles (C) in a curable resin composition containing the components (A) and (B) as main components, preferably polymer fine particles.
- (C) is a composition in which primary particles are dispersed.
- an aqueous latex containing polymer fine particles (C) (specifically, a reaction mixture after producing polymer fine particles by emulsion polymerization) has a solubility in water at 20 ° C. of 5% by mass.
- the first step of aggregating the polymer particles, and separating and collecting the agglomerated polymer fine particles (C) from the liquid phase A second step of mixing with an organic solvent to obtain an organic solvent solution of the polymer fine particles (C), and a third step of further distilling off the organic solvent after further mixing the organic solvent solution with the component (A). It is preferable to be prepared by.
- the component (A) is liquid at 23 ° C. because the third step is easy.
- “Liquid at 23 ° C.” means that the softening point is 23 ° C. or lower, and indicates fluidity at 23 ° C.
- each component of the present invention contains an isocyanate group-containing component ⁇ (B) component and / or an equivalent ratio (NCO / Active hydrogen-containing group), a first liquid containing an isocyanate group-containing urethane prepolymer obtained by reacting the component (A) and the component (B), a component having a hydroxyl group ⁇ the component (A), and / or Alternatively, as a second liquid containing a urethane prepolymer having a hydroxyl group obtained by reacting the component (A) and the component (B) at an equivalent ratio (NCO / active hydrogen-containing group) of less than 1, in a separate container It is preferable to store and mix before use.
- the components other than the component (A), the component (B), and the urethane prepolymer that is, the component (C), the component (D), and other compounding components are added to the first liquid.
- it may be added to the second liquid.
- two-component curing comprising a first liquid containing the component (B) and a second liquid containing the component (A), the component (C) and the component (D).
- the aspect made into the conductive resin composition is also included in the present invention.
- the present invention includes a cured product obtained by curing the curable resin composition.
- the polymer fine particles are dispersed in the form of primary particles. Therefore, by curing the polymer fine particles, a cured product in which the polymer fine particles are uniformly dispersed can be easily obtained.
- the polymer fine particles hardly swell and the viscosity of the curable resin composition is low, a cured product can be obtained with good workability.
- the glass transition temperature is preferably 75 ° C. or higher, more preferably 80 ° C. or higher, further preferably 90 ° C. or higher, still more preferably 100 ° C. or higher, and particularly preferably 110 ° C. or higher.
- the upper limit of the glass transition temperature is not particularly limited, but is about 200 ° C.
- the number of functional groups of the polyether polyol may be increased. For example, a tri- or higher functional polyether polyol, preferably a tetrafunctional polyether polyol is used.
- the amount of the tri- or higher functional polyether polyol is preferably 20% by mass or more, more preferably 30% by mass or more, still more preferably 40% by mass or more, and still more preferably 100% by mass of the polyol. It is 50 mass% or more, and it is preferable that it is 100 mass% or less, More preferably, it is 99 mass% or less, More preferably, it is 98 mass% or less, More preferably, it is 97 mass% or less. When the amount is less than 20% by mass, the glass transition temperature may not be improved.
- the amount of the tetrafunctional polyether polyol is preferably 50% by mass or more, more preferably 55% by mass or more, further preferably 60% by mass or more, and 90% by mass or less, in 100% by mass of the polyol. More preferably, it is 85 mass% or less, More preferably, it is 80 mass% or less.
- the amount is less than 50% by mass, the glass transition temperature may not be improved.
- the amount is more than 90% by mass, the rigidity becomes too high and the toughness may be lowered.
- the curable resin composition of the present invention includes structural adhesives, ink binders, wood chip binders, rubber chip binders, foam chip binders, foundry binders, floor materials, ceramics, bedrock binders, automotive interior materials, general woodworking, furniture. Adhesives for interiors, wall materials, food packaging, etc .; Coating materials; Fiber reinforced composite materials; Automobile seats, automotive interior parts, sound absorbing materials, damping materials, shock absorbers, heat insulating materials, construction flooring cushions, etc. It is preferably used for such applications.
- a structural adhesive or coating material using the curable resin composition of the present invention, and a curable resin composition of the present invention because it exhibits excellent toughness and adhesiveness while exhibiting high rigidity and high elastic modulus.
- a particularly preferred embodiment of the present invention includes a fiber-reinforced composite material obtained by using a product as a binder for reinforcing fibers.
- the curable resin composition of the present invention is excellent in adhesiveness and flexibility not only at a low temperature (about ⁇ 20 ° C.) to a normal temperature but also at a high temperature (about 80 ° C.). Therefore, the urethane resin adhesive composition of this embodiment can be preferably used as a structural adhesive.
- a laminate obtained by applying the curable resin composition of the present invention to a metal or a porous substrate and then curing the curable resin composition is excellent in anticorrosion to the substrate, while being resistant to cracking and resistance. Since it becomes a coating film excellent in loadability, it is included in the present invention as another embodiment.
- the application of the coating material using the curable resin composition of the present invention is not particularly limited, but includes automobiles, electrical equipment, office machines, building materials, wood, painted floors, heavy anticorrosion, concrete anticorrosion, roof / roof waterproofing and corrosion resistance.
- -Underground waterproofing coating film waterproofing material, electrodeposition paint, automobile repair can coating, top coat, intermediate coat, undercoat, primer, highly weather resistant paint, non-yellowing paint, etc.
- paint flooring, paving, etc. it can be used in factories, laboratories, warehouses, clean rooms, etc.
- the molding method of the composite material using the curable resin composition of the present invention is not particularly limited, but is an autoclave molding method using a prepreg, a filament wind molding method, a hand layup molding method, a vacuum bag molding method, a resin injection Examples include molding (RTM) method, vacuum assist resin injection molding (VARTM) method, pultrusion molding method, injection molding method, sheet winding molding method, spray-up method, BMC (Bulk Molding Compound) method, SMC (Sheet Molding Compound) method, etc. It is done.
- RTM molding
- VARTM vacuum assist resin injection molding
- pultrusion molding method injection molding method
- injection molding method sheet winding molding method
- spray-up method BMC (Bulk Molding Compound) method
- SMC Sheet Molding Compound
- the fracture toughness value K1c (MPa ⁇ m 1/2 ) was calculated according to the following formulas 2 and 3.
- a is the sum of the depth of the V notch and the length from the V notch tip to the crack tip, and L, h, a, and b are in cm.
- a graft monomer 60 parts by mass of methyl methacrylate (MMA), 30 parts by mass of 4-hydroxybutyl acrylate (4HBA)), and cumene hydro
- CHP peroxide
- Production Example 2-2 Preparation of core-shell polymer latex (L-2) A glass reactor having a thermometer, a stirrer, a reflux condenser, a nitrogen inlet, and a monomer addition apparatus was obtained in Production Example 1-2. 240 parts by mass of polybutadiene rubber latex (R-2) (including 80 parts by mass of polybutadiene rubber particles) and 57 parts by mass of deionized water were charged and stirred at 60 ° C. while purging with nitrogen.
- R-2 polybutadiene rubber latex
- Production Example 2-4 Preparation of Core Shell Polymer Latex (L-4)
- Production Example 2-2 instead of ⁇ MMA 18 parts by mass, styrene (ST) 2 parts by mass> as a graft monomer, ⁇ MMA 17 parts by mass, ST 2 parts by mass
- a latex (L-4) of the core-shell polymer (C-4) was obtained in the same manner as in Production Example 2-2 except that 1 part by mass of 4HBA was used.
- the volume average particle diameter of the core-shell polymer contained in the obtained latex was 0.21 ⁇ m.
- Production Example 2-5 Preparation of Core Shell Polymer Latex (L-5)
- Production Example 2-2 instead of ⁇ MMA 18 parts by mass, styrene (ST) 2 parts by mass> as a graft monomer, ⁇ MMA 16 parts by mass, ST 2 parts by mass
- a latex (L-5) of the core-shell polymer (C-5) was obtained in the same manner as in Production Example 2-2 except that 4 parts by mass of 4HBA was used.
- the volume average particle diameter of the core-shell polymer contained in the obtained latex was 0.21 ⁇ m.
- Production Example 2-6 Preparation of Core Shell Polymer Latex (L-6)
- Production Example 2-2 instead of ⁇ MMA 18 parts by mass, styrene (ST) 2 parts by mass> as a graft monomer, ⁇ MMA 14 parts by mass, ST 2 parts by mass
- a latex (L-6) of the core-shell polymer (C-6) was obtained in the same manner as in Production Example 2-2 except that 4 parts by mass of 4HBA was used.
- the volume average particle diameter of the core-shell polymer contained in the obtained latex was 0.21 ⁇ m.
- a slurry liquid composed of an aqueous phase partially containing a floating aggregate and an organic solvent was obtained.
- an agglomerate containing a part of the aqueous phase was left, and 360 g of the aqueous phase was discharged from the discharge port at the bottom of the tank.
- 90 g of MEK was added to the obtained aggregate and mixed uniformly to obtain a dispersion in which the core-shell polymer was uniformly dispersed.
- 80 g of trifunctional polyether polyol PPT300 (A-1: Actol T-300, manufactured by Mitsui Chemicals) as component (A) was mixed. From this mixture, MEK was removed with a rotary evaporator.
- a dispersion (M-1) in which polymer fine particles were dispersed in component (A) was obtained.
- Production Example 3-3 Preparation of Dispersion (M-3) In Production Example 3-1, instead of PPT300 (A-1), a bifunctional polyether polyol PPG400 (A-3, manufactured by Mitsui Chemicals, Actol D) A dispersion (M-3) in which polymer fine particles were dispersed was obtained in the same manner as in Production Example 3-1, except that -400) was used.
- Example 2 and Comparative Examples 1 and 2 were hard cured plates, the bending strain and the fracture toughness K1c at the maximum bending stress were measured using the cured plates according to the test method described above. .
- the test results are shown in Table 1.
- Example 2 and Comparative Example 2 unstable fracture did not occur when measuring fracture toughness, and an effective value of K1c was not obtained.
- the cured plates of Comparative Examples 3 to 4 were soft cured plates, it was not possible to measure bending physical properties and fracture toughness. Therefore, in accordance with JIS K 6251, a No. 3 dumbbell test piece was taken from the cured plate and subjected to a tensile test at 23 ° C. at a speed of 500 mm / min, and the elongation at the maximum tensile stress was measured. The test results are shown in Table 1.
- the cured product obtained by curing the curable resin composition of the present invention exhibits excellent fracture toughness. Moreover, when the polyether polyol (a1), polyisocyanate (B), and polymer fine particles (C) having a specific average hydroxyl value are combined from the values of bending strain and tensile elongation, high ductility and high elongation characteristics are exhibited. I understand that.
- Table 2 shows that the curable resin composition of the present invention has a high elastic modulus (high rigidity) and excellent fracture toughness.
- component which is aromatic polyisocyanate which has a cyclic structure is used, the fracture toughness improvement effect by a combination with polymer fine particles (C) is remarkable.
- (A) component has much content of polyfunctional polyol more than trifunctional, it turns out that high heat resistance (a glass transition temperature is high) is shown.
- the content of the polyfunctional polyol having a functionality of 3 or more among the component (A) is large, the effect of improving the fracture toughness by the combination with the polymer fine particles (C) is remarkable.
- a dehydrating agent (powdered synthetic zeolite A-3 (200 mesh passage: manufactured by Wako Pure Chemical Industries) and silica as a filler (manufactured by Tatsumori, Crystallite C) were added and mixed well.
- the dehydrating agent was used, it was used as it was in the next step without removing the dehydrating agent, and the polyol-silica mixture was then used as the component (B) polymer MDI (B-2: manufactured by Mitsui Chemicals).
- this composition was poured into a forming die made of a Teflon (registered trademark) coated steel plate whose temperature was adjusted to 40 ° C. to obtain a foamed molded product having a thickness of 5 mm.
- the molding density was measured from the weight of the foamed molded product, the apparent volume, and the specific gravity of the unfoamed molded product. Further, Charpy strength was measured using this foamed molded article in accordance with JIS K7111-1. The test results are shown in Table 4.
- the obtained mixed solution was thoroughly mixed with polymer MDI (B-2: manufactured by Mitsui Chemicals, Cosmonate M-200) as component (B) to obtain a curable resin composition.
- polymer MDI B-2: manufactured by Mitsui Chemicals, Cosmonate M-200
- the value ( ⁇ / ⁇ ) of the ratio between the total molar amount ( ⁇ ) of the isocyanate group of the component (B) and the total molar amount ( ⁇ ) of the hydroxyl group of the component (A) in the composition of Table 5 is all 1.1.
- This composition was sucked into a mold in which eight carbon fiber fabrics of 195 g / m 2 were stacked, and then heated at 100 ° C. for 3 hours to obtain a carbon fiber reinforced molded body.
- the ratio values ( ⁇ / ⁇ ) of the total molar amount ( ⁇ ) of the isocyanate group of the component (B) in the composition of Table 6 and the total molar amount ( ⁇ ) of the hydroxyl group of the component (A) are all 1.1.
- the T-shaped peel strength was measured according to the test method described above. The test results are shown in Table 6.
- the value ( ⁇ / ⁇ ) of the ratio between the total molar amount ( ⁇ ) of the isocyanate group of the component (B) and the total molar amount ( ⁇ ) of the hydroxyl group of the component (A) in the composition of Table 7 is all 1.1.
- T-peel strength and dynamic splitting resistance (impact peel adhesion) were measured. The test results are shown in Table 7.
- the curable resin composition of the present invention exhibits high T-peeling strength and high dynamic splitting resistance. It turns out that it is excellent in adhesiveness.
- Example 18 Using the curable resin composition of Example 1, it apply
- Example 19 A curable resin obtained by changing 1 part by mass of DBU octylate (San Apro: U-CAT SA102), which is the component (D), of the curable resin composition of Example 1 to 0.01 parts by mass of dibutyltin dilaurate Using the composition, it was applied in a thickness of 100 ⁇ m on a mortar board in the same manner as in Example 18. This was cured at 80 ° C. for 3 hours to obtain a laminate. The coating film of the obtained laminate was a coating film resistant to impact. Furthermore, the coating film of this laminate was left in a sunny place outside the room for one year, but no yellowing of the coating film was observed.
- DBU octylate San Apro: U-CAT SA102
- D dibutyltin dilaurate
- Example 20 Using the curable resin composition of Example 1, a glass fiber reinforced composite material was produced according to the example of JP-T-2002-530445. The resulting composite material exhibited high toughness.
Abstract
Description
また、本発明の目的は、前記硬化性樹脂組成物を用いた技術を提供することにもある。
[1] 平均水酸基価が200~1500mgKOH/gであるポリオール(A)と、ポリイソシアネート(B)と、ポリマー微粒子(C)を含有することを特徴とする硬化性樹脂組成物。
[2] 前記ポリオール(A)として、ポリエーテルポリオール(a1)を含有することを特徴とする[1]に記載の硬化性樹脂組成物。
[3] 前記ポリオール(A)の平均水酸基価が350~1500mgKOH/gであり、さらに硬化触媒(D)を含有することを特徴とする[1]または[2]に記載の硬化性樹脂組成物。
[4] 前記ポリエーテルポリオール(a1)の量が、ポリオール(A)成分の総量100質量部の内、50質量部以上であることを特徴とする[2]または[3]に記載の硬化性樹脂組成物。
[5] ポリオール(A)と、ポリイソシアネート(B)と、ポリマー微粒子(C)を含有し、前記ポリオール(A)として、ポリエステルポリオール(a2)を必ず含有し、かつポリエステルポリオール(a2)の量が、ポリオール(A)成分の総量100質量部の内、20質量部以上であることを特徴とする硬化性樹脂組成物。
[6] 前記ポリオール(A)の平均水酸基価が20~1000mgKOH/gであることを特徴とする[5]に記載の硬化性樹脂組成物。
[7] さらに硬化触媒(D)を含有する[5]または[6]に記載の硬化性樹脂組成物。
[8] 前記ポリイソシアネート(B)を、前記ポリオール(A)100質量部に対して、2~5000質量部含有し、
前記ポリマー微粒子(C)を、前記ポリオール(A)と前記ポリイソシアネート(B)の総量100質量部に対して、1~100質量部含有することを特徴とする[1]~[7]のいずれかに記載の硬化性樹脂組成物。
[9] 前記(C)成分の体積平均粒子径が、10~2000nmであることを特徴とする[1]~[8]のいずれかに記載の硬化性樹脂組成物。
[10] 前記(C)成分が、コアシェル構造を有することを特徴とする[1]~[9]のいずれかに記載の硬化性樹脂組成物。
[11] 前記(C)成分が、ジエン系ゴム、(メタ)アクリレート系ゴム、及びオルガノシロキサン系ゴムよりなる群から選択される1種以上のコア層を有することを特徴とする[1]~[10]のいずれかに記載の硬化性樹脂組成物。
[12] 前記ジエン系ゴムが、ブタジエンゴム、およびブタジエン-スチレンゴムよりなる群から選択される1種以上であることを特徴とする[11]に記載の硬化性樹脂組成物。
[13] 前記(C)成分が、芳香族ビニルモノマー、ビニルシアンモノマー、及び(メタ)アクリレートモノマーよりなる群から選択される1種以上のモノマー成分を、コア層にグラフト重合してなるシェル層を有することを特徴とする[1]~[12]のいずれかに記載の硬化性樹脂組成物。
[14] 前記(C)成分が、水酸基を有するモノマー成分を、コア層にグラフト重合してなるシェル層を有することを特徴とする[1]~[13]のいずれかに記載の硬化性樹脂組成物。
[15] 前記(C)成分が、該硬化性樹脂組成物中で1次粒子の状態で分散していることを特徴とする[1]~[14]のいずれかに記載の硬化性樹脂組成物。
[16] 前記(A)成分が、3官能以上の多官能ポリオールを含有することを特徴とする[1]~[15]のいずれかに記載の硬化性樹脂組成物。
[17] 前記(A)成分の総量100質量部の内、3官能以上の多官能ポリオールの含有量が20質量部以上であることを特徴とする[16]に記載の硬化性樹脂組成物。
[18] 前記(B)成分が、環状構造または直鎖構造もしくは分岐鎖構造を有するポリイソシアネートであることを特徴とする[1]~[17]のいずれかに記載の硬化性樹脂組成物。
[19] 前記(B)成分が、芳香族ポリイソシアネートであることを特徴とする[18]に記載の硬化性樹脂組成物。
[20] 前記(B)成分が、イソシアネート基を1分子当り平均して2.1個以上有するポリイソシアネートであることを特徴とする[1]~[19]のいずれかに記載の硬化性樹脂組成物。
[21] 前記(B)成分が、脂環族ポリイソシアネート、および脂肪族ポリイソシアネートよりなる群から選択される1種以上であることを特徴とする[18]に記載の硬化性樹脂組成物。
[22] 前記(B)成分が、脂環族ポリイソシアネートである[21]に記載の硬化性樹脂組成物。
[23] 前記(A)成分と前記(B)成分の総量100質量部に対して、硬化触媒(D)0.001~20質量部をさらに含有することを特徴とする[1]~[22]のいずれかに記載の硬化性樹脂組成物。
[24] 前記(D)成分が有機スズ化合物であることを特徴とする[23]に記載の硬化性樹脂組成物。
[25] 前記(A)成分と前記(B)成分の総量100質量部に対して、脱水剤(E)0.1~30質量部をさらに含有することを特徴とする[1]~[24]のいずれかに記載の硬化性樹脂組成物。
[26] 前記(B)成分のイソシアネート基の総モル量(β)と前記(A)成分の水酸基の総モル量(α)との比の値(β/α)が、0.7~1.5であることを特徴とする[1]~[25]のいずれかに記載の硬化性樹脂組成物。
[27] 前記(A)成分と前記(B)成分との反応により得られるウレタンプレポリマーを含有することを特徴とする[1]~[26]のいずれかに記載の硬化性樹脂組成物。
[28] 前記(B)成分のイソシアネート(NCO)基と前記(A)成分の活性水素含有基との当量比(NCO/活性水素含有基)が、1.05~5.0の範囲で反応して得たイソシアネートを有するウレタンプレポリマーを含有することを特徴とする[27]に記載の硬化性樹脂組成物。
[29] [28]に記載の硬化性樹脂組成物からなる1液型湿分硬化性樹脂組成物。
[30] [1]~[28]のいずれかに記載の硬化性樹脂組成物であって、前記(B)成分を含有する第1液と、前記(A)成分と前記(C)成分と前記(D)成分を含有する第2液とからなる2液型硬化性樹脂組成物。
[31] [1]~[30]のいずれかに記載の硬化性樹脂組成物を硬化して得られる硬化物。
[32] [1]~[30]のいずれかに記載の硬化性樹脂組成物を用いてなる構造接着剤。
[33] [1]~[30]のいずれかに記載の硬化性樹脂組成物を用いてなるコーティング材。
[34] [1]~[30]のいずれかに記載の硬化性樹脂組成物を、金属または多孔質下地に塗布した後、該硬化性樹脂組成物を硬化してなる積層体。
[35] [1]~[30]のいずれかに記載の硬化性樹脂組成物を、強化繊維のバインダーとして用いてなる繊維強化複合材料。
[36] [1]~[30]のいずれかに記載の硬化性樹脂組成物を発泡してなる発泡体。
[37] ガラス転移温度が75℃以上である[31]に記載の硬化物。
第一の態様は、平均水酸基価が200~1500mgKOH/gであるポリオール(A)と、ポリイソシアネート(B)と、ポリマー微粒子(C)を含有することを特徴とする硬化性樹脂組成物(I)である。
第二の態様は、ポリオール(A)と、ポリイソシアネート(B)と、ポリマー微粒子(C)を含有し、前記ポリオール(A)として、ポリエステルポリオールを必ず含有し、ポリエステルポリオール(a2)の量が、ポリオール(A)成分の総量100質量部の内、20質量部以上であることを特徴とする硬化性樹脂組成物(II)である。
前記第一の態様及び第二の態様のいずれでも、さらに硬化触媒(D)を含有していてもよい。
第一の態様は、特に従来よりも高い剛性(弾性率)及び靭性を示す点で有利であり、第二の態様は、特に従来と比較して同等以上の接着性を示す点で有利である。
本発明の硬化性樹脂組成物では、ポリオール(A)を使用する。ポリオール(A)は単独で用いても良く2種以上併用しても良い。
前記の3官能以上の多官能ポリオールの含有量は、前記(A)成分の全量(総量100質量部)の内、20質量部以上が好ましく、30質量部以上がより好ましく、50質量部以上が更に好ましく、70質量部以上が特に好ましい。前記の3官能以上の多官能ポリオールの含有量は、例えば100質量部以下であり、好ましくは90質量部以下、より好ましくは80質量部以下である。20質量部未満では、得られる硬化物の剛性が不十分な場合がある。
また、該硬化性樹脂組成物から無機成分を除いた全質量に対して、(A)成分の含有量は、25質量%以上が好ましく、30質量%以上がより好ましく、35質量%以上が更に好ましく、40質量%以上が特に好ましい。上記(A)成分の含有量は、該硬化性樹脂組成物から無機成分を除いた全質量に対して、75質量%以下が好ましく、より好ましくは70質量%以下、更に好ましくは65質量%以下、特に好ましくは60質量%以下である。25質量%未満または75質量%超では、得られる硬化物の靱性と接着性が低下する場合がある。
本発明の硬化性樹脂組成物では、ポリイソシアネート(B)を使用する。ポリイソシアネート(B)は単独で用いても良く2種以上併用しても良い。ポリイソシアネート(B)は、本発明の(A)成分と反応し、硬化性樹脂組成物中のポリウレタンを形成する必須構成成分である。
また、該硬化性樹脂組成物から無機充填材やガラス繊維・炭素繊維等の無機成分を除いた全質量(例えば、(A)~(E)成分の全質量)に対して、(B)成分の含有量は、25質量%以上が好ましく、30質量%以上がより好ましく、35質量%以上が更に好ましく、40質量%以上が特に好ましい。上記(B)成分の含有量は、該硬化性樹脂組成物の全量の70質量%以下が好ましく、より好ましくは65質量%以下、更に好ましくは60質量%以下、特に好ましくは55質量%以下である。25質量%未満または70質量%超では、得られる硬化物の靱性と接着性が低下する場合がある。
本発明では、硬化性樹脂組成物の硬化時に初めて(A)成分と(B)成分を反応させることも可能であるが、(A)成分と(B)成分の一部または全部を事前に反応させた所謂プレポリマーとして、硬化性樹脂組成物に使用することも可能である。さらには、(C)成分を(A)成分に分散させてから、(B)成分と反応させてプレポリマー化してもよい。この様に、前記硬化性樹脂組成物は、前記(A)成分と前記(B)成分との反応により得られるウレタンプレポリマーを含有してもよい。プレポリマー化により、ウレタン化反応の反応性制御、2液型硬化性樹脂組成物の場合の混合比制御、硬化性樹脂組成物の粘度調節、硬化時の発泡抑制などの効果がある。
本発明の硬化性樹脂組成物は、前記(A)成分と前記(B)成分の総量100質量部に対して、ポリマー微粒子(C)1~100質量部を使用することが好ましい。(C)成分の添加により、得られる硬化物は靱性、耐クラック性、および、接着性に優れる。
ポリマー微粒子の分散状態は、例えば、硬化性樹脂組成物の一部をメチルエチルケトンのような溶剤に溶解し、これをレーザー光散乱による粒子径測定装置等により、その粒子径を測定することにより確認できる。または、硬化性樹脂組成物を硬化させた後、透過型電子顕微鏡(TEM)または走査型電子顕微鏡(SEM)を用いて観察すれば、容易に確認できる。ポリマー微粒子が組成物中で凝集している場合、粒子の凝集力が非常に強いため、組成物を溶剤で希釈しても凝集体を一次粒子に分離することはできない。また、硬化前の組成物においてポリマー微粒子が一次分散していないにもかかわらず、硬化後にポリマー微粒子が一次分散する可能性はなく、硬化物中においてポリマー微粒子が一次分散していれば、硬化前の組成物でもポリマー微粒子は一次分散している。
本発明において、ポリマー微粒子は、硬化性樹脂組成物またはその硬化物中に、粉体のポリマー微粒子または凝固・乾燥されたポリマー微粒子を含まないことが好ましく、後述の通り、水性ラテックスに由来し、かつ凝固・乾燥されていないポリマー微粒子からなることが好ましい。かかるポリマー微粒子であれば、一次粒子の分散状態を容易に実現することができ、一方で粉体のポリマー微粒子であれば、一次粒子の分散状態を実現することが困難となる。
(なお、(式1)において、B1は、3個以上が接触しているゴム状ポリマー粒子(B)の個数B1(なお、ある1個のゴム状ポリマー粒子(B)がn個に接触している場合、個数はn個とカウントする)であり、B0は、ゴム状ポリマー粒子(B)の総個数である。)
≪コア層≫
コア層は、本発明の硬化性樹脂組成物の硬化物の靱性を高める為に、ゴムとしての性質を有する弾性コア層であることが好ましい。ゴムとして性質を有するためには、本発明の弾性コア層は、ゲル含量が60質量%以上であることが好ましく、80質量%以上であることがより好ましく、90質量%以上であることがさらに好ましく、95質量%以上であることが特に好ましい。なお、本明細書でいうゲル含量とは、凝固、乾燥により得られたポリマー微粒子0.5gをトルエン100gに浸漬し、23℃で24時間静置した後に不溶分と可溶分を分別したときの、不溶分と可溶分の合計量に対する不溶分の比率を意味する。
弾性コア層に用いるジエン系ゴムを構成するモノマー(共役ジエン系モノマー)としては、例えば、1,3-ブタジエン、イソプレン、2-クロロ-1,3-ブタジエン、2-メチル-1,3-ブタジエンなどが挙げられる。これらのジエン系モノマーは、単独で用いても、2種以上を組み合わせて用いてもよい。
靱性改良効果の点から、1,3-ブタジエンを用いるブタジエンゴム、または、1,3-ブタジエンとスチレンの共重合体であるブタジエン-スチレンゴムが好ましく、ブタジエンゴムがより好ましい。また、ブタジエン-スチレンゴムは、屈折率の調整により得られる硬化物の透明性を高めることができ、より好ましい。
本発明では、必要により、中間層を形成させてもよい。特に、中間層として、以下のゴム表面架橋層を形成させてもよい。
ポリマー微粒子の最も外側に存在するシェル層は、シェル形成用単量体を重合したものであるが、本発明に係る、ポリマー微粒子(C)と(A)成分あるいは(B)成分との相溶性を向上させ、本発明の硬化性樹脂組成物、又はその硬化物中においてポリマー微粒子が一次粒子の状態で分散することを可能にする役割を担うシェルポリマーからなる。
すなわち、前記(C)成分は、芳香族ビニルモノマー、ビニルシアンモノマー、及び(メタ)アクリレートモノマーよりなる群から選択される1種以上のモノマー成分を、コア層にグラフト重合してなるシェル層を有することが好ましく、前記(C)成分は、水酸基を有するモノマー成分を、コア層にグラフト重合してなるシェル層を有することが好ましい。
(コア層の製造方法)
本発明で用いるポリマー微粒子を構成するコア層を形成するポリマーが、ジエン系モノマー(共役ジエン系モノマー)および(メタ)アクリレート系モノマーから選ばれる少なくとも1種のモノマー(第1モノマー)を含んで構成される場合には、コア層の形成は、例えば、乳化重合、懸濁重合、マイクロサスペンジョン重合などによって製造することができ、例えばWO2005/028546号公報に記載の方法を用いることができる。
中間層は、中間層形成用モノマーを公知のラジカル重合により重合することによって形成することができる。コア層を構成するゴム弾性体をエマルジョンとして得た場合には、二重結合を2以上有するモノマーの重合は乳化重合法により行うことが好ましい。
本発明の硬化性樹脂組成物には必要に応じて(D)成分として硬化触媒を使用することができる。硬化触媒としては、特に限定されず、通常使用されるウレタン化反応を促進する触媒が挙げられる。具体例としては、2-エチルヘキサン酸錫、バーサチック酸錫、2-エチルヘキサン酸ビスマス、酢酸カリウム、オクチル酸カリウム、オクチル酸鉛、ナフテン酸鉛、ナフテン酸ニッケル、オクチル酸コバルト等のカルボン酸金属塩;ジブチル錫ジラウレート、ジブチル錫マレエート、ジブチル錫フタレート、ジブチル錫ジオクタノエート、ジブチル錫ビス(2-エチルヘキサノエート)、ジブチル錫ビス(メチルマレエート)、ジブチル錫ビス(エチルマレエート)、ジブチル錫ビス(ブチルマレエート)、ジブチル錫ビス(オクチルマレエート)、ジブチル錫ビス(トリデシルマレエート)、ジブチル錫ビス(ベンジルマレエート)、ジブチル錫ジアセテート、ジオクチル錫ビス(エチルマレエート)、ジオクチル錫ビス(オクチルマレエート)、ジブチル錫ジメトキサイド、ジブチル錫ビス(ノニルフェノキサイド)、ジブテニル錫オキサイド、ジブチル錫ビス(アセチルアセトナート)、ジブチル錫ビス(エチルアセトアセテート)、ジブチル錫ビスイソオクチルチオグリコレート、ジブチル錫ジクロライド、ジブチル錫オキサイド、ジブチル錫オキサイドとシリケート化合物との反応物、ジブチル錫ジラウレート等のジアルキル錫ジカルボキシレートとシリケート化合物との反応物、ジブチル錫オキサイドとフタル酸エステルとの反応物等の4価の有機錫化合物;テトライソプロポキシチタニウム、テトラn-ブトキシチタニウム、ジイソプロポキシチタンビス(アセチルアセトナート)、ジイソプロポキシチタンビス(エチルアセトアセテート)などの有機チタネート類;アルミニウムトリス(アセチルアセトナート)、アルミニウムトリス(エチルアセトアセテート)、ジイソプロポキシアルミニウムエチルアセトアセテートなどの有機アルミニウム化合物類;ジルコニウムテトラキス(アセチルアセトナート)などのジルコニウム化合物類;トリアミルアミン、トリヘキシルアミン、トリオクチルアミン、トリアリルアミン、トリフェニルアミン、トリエタノールアミン、トリエチルアミン、トリプロピルアミン、ジエチルエタノールアミン、ジメチルアミノエトキシエタノール、N,N,N’-トリメチルアミノエチルエタノールアミン、N,N,N’,N”,N”-ペンタメチルジエチレントリアミン、N,N,N’,N’-テトラメチルヘキサメチレンジアミン、N,N,N’,N’-テトラメチルエチレンジアミン、N,N’,N’-トリメチルアミノエチルピペラジン、N,N-ジメチルシクロヘキシルアミン、ビス(2-ジメチルアミノエチル)エーテル、グアニジン、ジフェニルグアニジン、2,4,6-トリス(ジメチルアミノメチル)フェノール、N-メチルモルホリン、N-エチルモルホリン、N-オクタデシルモルホリン、N-メチルピペラジン、N-メチル-N’-(2-ヒドロキシプロピル)ピペラジン、2-エチル-4-メチルイミダゾール、1,8-ジアザビシクロ(5,4,0)ウンデセン(DBU)、1,5-ジアザビシクロ(4,3,0)ノネン(DBN)、1,4-ジアザビシクロ[2.2.2]オクタン(DABCO)などのアミン類化合物等が挙げられる。中でも、前記(D)成分は、黄変抑制の観点から、有機スズ化合物であることが好ましい。また、DBUのオクチル酸塩など、前記アミン類化合物とカルボン酸やフェノール類などの有機酸との塩は、潜在硬化性触媒として有効である。
本発明の硬化性樹脂組成物には、充填材を添加することができる。充填材としては、フュームドシリカ、沈降性シリカ、結晶性シリカ、溶融シリカ、ドロマイト、無水ケイ酸、含水ケイ酸、およびカーボンブラックの如き補強性充填材;重質炭酸カルシウム、膠質炭酸カルシウム、炭酸マグネシウム、炭酸バリウム、硫酸バリウム、ケイソウ土、焼成クレー、クレー、タルク、バライト、無水石膏、酸化チタン、ベントナイト、有機ベントナイト、酸化第二鉄、アルミニウム微粉末、フリント粉末、酸化亜鉛、活性亜鉛華、マイカ、亜鉛華、鉛白、リトポン、硫化亜鉛、シラスバルーン、ガラスミクロバルーン、フェノール樹脂や塩化ビニリデン樹脂の有機ミクロバルーン、PVC粉末、PMMA粉末など樹脂粉末の如き充填材;石綿、ガラス繊維およびフィラメントの如き繊維状充填材等が挙げられる。
本発明の硬化性樹脂組成物には、必要に応じて、ポリリン酸アンモニウム、トリクレジルホスフェートなどのリン系可塑剤、水酸化アルミニウム、水酸化マグネシウム、および、熱膨張性黒鉛などの難燃剤を添加することができる。上記難燃剤は単独で用いてもよく、2種以上併用してもよい。
本発明の硬化性樹脂組成物には、必要に応じて分散剤を添加することができる。該分散剤は、顔料とともに公知の方法に従って混合分散して得られる顔料分散ペーストを配合することも可能である。分散剤としては、市販されているものを使用することができる。市販品としては、例えば、ANTI-TERRA(登録商標)-U、ANTI-TERRA(登録商標)-U100、ANTI-TERRA(登録商標)-204、ANTI-TERRA(登録商標)-205、DISPERBYK(登録商標)-101、DISPERBYK(登録商標)-102、DISPERBYK(登録商標)-103、DISPERBYK(登録商標)-106、DISPERBYK(登録商標)-108、DISPERBYK(登録商標)-109、DISPERBYK(登録商標)-110、DISPERBYK(登録商標)-111、DISPERBYK(登録商標)-112、DISPERBYK(登録商標)-116、DISPERBYK(登録商標)-130、DISPERBYK(登録商標)-140、DISPERBYK(登録商標)-142、DISPERBYK(登録商標)-145、DISPERBYK(登録商標)-161、DISPERBYK(登録商標)-162、DISPERBYK(登録商標)-163、DISPERBYK(登録商標)-164、DISPERBYK(登録商標)-166、DISPERBYK(登録商標)-167、DISPERBYK(登録商標)-168、DISPERBYK(登録商標)-170、DISPERBYK(登録商標)-171、DISPERBYK(登録商標)-174、DISPERBYK(登録商標)-180、DISPERBYK(登録商標)-182、DISPERBYK(登録商標)-183、DISPERBYK(登録商標)-184、DISPERBYK(登録商標)-185、DISPERBYK(登録商標)-2000、DISPERBYK(登録商標)-2001、DISPERBYK(登録商標)-2008、DISPERBYK(登録商標)-2009、DISPERBYK(登録商標)-2022、DISPERBYK(登録商標)-2025、DISPERBYK(登録商標)-2050、DISPERBYK(登録商標)-2070、DISPERBYK(登録商標)-2096、DISPERBYK(登録商標)-2150、DISPERBYK(登録商標)-2155、DISPERBYK(登録商標)-2163、DISPERBYK(登録商標)-2164、BYK(登録商標)-P104、BYK(登録商標)-P104S、BYK(登録商標)-P105、BYK(登録商標)-9076、BYK(登録商標)-9077、BYK(登録商標)-220S、ANTI-TERRA(登録商標)-250、DISPERBYK(登録商標)-187、DISPERBYK(登録商標)-190、DISPERBYK(登録商標)-191、DISPERBYK(登録商標)-192、DISPERBYK(登録商標)-193、DISPERBYK(登録商標)-194、DISPERBYK(登録商標)-198、DISPERBYK(登録商標)-2010、DISPERBYK(登録商標)-2012、DISPERBYK(登録商標)-2015、DISPERBYK(登録商標)-2090、DISPERBYK(登録商標)-2091、DISPERBYK(登録商標)-2095(いずれもビックケミー社製)、DISPARLON(登録商標) 2150、DISPARLON(登録商標) KS-860、DISPARLON(登録商標) KS-873N、DISPARLON(登録商標) 7004、DISPARLON(登録商標) 1831、DISPARLON(登録商標) 1850、DISPARLON(登録商標) 1860、DISPARLON(登録商標) DA-1401、DISPARLON(登録商標) PW-36、DISPARLON(登録商標) DA-1200、DISPARLON(登録商標) DA-550、DISPARLON(登録商標) DA-703-50、DISPARLON(登録商標) DA-7301、DISPARLON(登録商標) DN-900、DISPARLON(登録商標) DA-325、DISPARLON(登録商標) DA-375、DISPARLON(登録商標) DA-234(いずれも楠本化成社製)、EFKAPOLYMER4550(EFKA社製)、ソルスパース(登録商標)27000、ソルスパース(登録商標)41000、ソルスパース(登録商標)53095(いずれもアビシア社製)等を挙げることができる。これらの中でも、ANTI-TERRA(登録商標)-U100、DISPERBYK(登録商標)-102、DISPERBYK(登録商標)-106、DISPERBYK(登録商標)-108、DISPERBYK(登録商標)-109、DISPERBYK(登録商標)-111、DISPERBYK(登録商標)-116、DISPERBYK(登録商標)-145、DISPERBYK(登録商標)-180、DISPERBYK(登録商標)-185、DISPERBYK(登録商標)-2008、DISPERBYK(登録商標)-2096、DISPERBYK(登録商標)-2155、BYK(登録商標)-P105、BYK(登録商標)-9076、BYK(登録商標)-9077、DISPERBYK(登録商標)-191、DISPERBYK(登録商標)-192、DISPERBYK(登録商標)-2090、DISPERBYK(登録商標)-2095、DISPARLON(登録商標) DA-550、DISPARLON(登録商標) DA-325、DISPARLON(登録商標) DA-375、DISPARLON(登録商標) DA-234は不揮発分含量が高く好ましい。
本発明の硬化性樹脂組成物には、必要に応じて消泡剤を添加することができる。消泡剤としては、市販されているものを使用することができる。市販品としては、例えば、BYK(登録商標)-051、BYK(登録商標)-052、BYK(登録商標)-053、BYK(登録商標)-054、BYK(登録商標)-055、BYK(登録商標)-057、BYK(登録商標)-1752、BYK(登録商標)-1790、BYK(登録商標)-060N、BYK(登録商標)-063、BYK(登録商標)-065、BYK(登録商標)-066N、BYK(登録商標)-067A、BYK(登録商標)-077、BYK(登録商標)-088、BYK(登録商標)-141、BYK(登録商標)-354、BYK(登録商標)-392、BYK(登録商標)-011、BYK(登録商標)-012、BYK(登録商標)-017、BYK(登録商標)-018、BYK(登録商標)-019、BYK(登録商標)-020、BYK(登録商標)-021、BYK(登録商標)-022、BYK(登録商標)-023、BYK(登録商標)-024、BYK(登録商標)-025、BYK(登録商標)-028、BYK(登録商標)-038、BYK(登録商標)-044、BYK(登録商標)-093、BYK(登録商標)-094、BYK(登録商標)-1610、BYK(登録商標)-1615、BYK(登録商標)-1650、BYK(登録商標)-1730、BYK(登録商標)-1770などの消泡剤(いずれもビックケミー社製)や、DISPARLON(登録商標) OX-880EF、DISPARLON(登録商標) OX-881、DISPARLON(登録商標) OX-883、DISPARLON(登録商標) OX-883HF、DISPARLON(登録商標) OX-70、DISPARLON(登録商標) OX-77EF、DISPARLON(登録商標) OX-60、DISPARLON(登録商標) OX-710、DISPARLON(登録商標) OX-720、DISPARLON(登録商標)
OX-720EF、DISPARLON(登録商標) OX-750HF、DISPARLON(登録商標) LAP-10、DISPARLON(登録商標) LAP-20、DISPARLON(登録商標) LAP-30等のアクリル系消泡剤、DISPARLON(登録商標) OX-66、DISPARLON(登録商標) OX-715等のシリコーン系アクリル系複合型消泡剤、DISPARLON(登録商標) 1950、DISPARLON(登録商標) 1951、DISPARLON(登録商標) 1952、DISPARLON(登録商標) P-410EF、DISPARLON(登録商標) P-420、DISPARLON(登録商標) P-450、DISPARLON(登録商標) P-425、DISPARLON(登録商標) PD-7等のビニル系消泡剤、DISPARLON(登録商標) 1930N、DISPARLON(登録商標) 1934等のシリコーン系消泡剤(いずれも楠本化成社製)等を挙げることができる。
本発明の硬化性樹脂組成物には、必要に応じて可塑剤を添加することができる。可塑剤の添加により、硬化性樹脂組成物の粘度やスランプ性および組成物を硬化して得られる硬化物の引張り強度、伸びなどの機械特性が調整できる。可塑剤の例としては、ジブチルフタレート、ジヘプチルフタレート、ジ(2-エチルヘキシル)フタレート、ブチルベンジルフタレート等のフタル酸エステル類;ジオクチルアジペート、ジオクチルセバケート、ジブチルセバケート、コハク酸イソデシル等の非芳香族二塩基酸エステル類;ジエチレングリコールベンゾエート、ジペンタエリスリトールへキサエステル等のグリコールエステル類;オレイン酸ブチル、アセチルリシリノール酸メチル等の脂肪族エステル類;トリクレジルホスフェート、トリブチルホスフェート等のリン酸エステル類;トリメリット酸エステル類;塩素化パラフィン類;アルキルジフェニル、部分水添ターフェニル等の炭化水素系油;プロセスオイル類;エポキシ化大豆油、エポキシステアリン酸ベンジル等のエポキシ可塑剤類をあげることができる。
本発明の硬化性樹脂組成物には、組成物の粘度を低減し、チクソ性を高め、作業性を改善する目的で、必要に応じて溶剤を使用することができる。溶剤としては、特に限定は無く、各種の化合物を使用することができる。具体例としては、トルエン、キシレン、ヘプタン、ヘキサン、石油系溶媒等の炭化水素系溶剤、トリクロロエチレン等のハロゲン系溶剤、酢酸エチル、酢酸ブチル等のエステル系溶剤、アセトン、メチルエチルケトン、メチルイソブチルケトン等のケトン系溶剤、ジブチルエーテル、ジペンチニルエーテル、ジエチレングリコールモノメチルエーテル、ジエチレングリコールモノエチルエーテル等のエーテル系溶剤、ヘキサメチルシクロトリシロキサン、オクタメチルシクロテトラシロキサン、デカメチルシクロペンタシロキサン等のシリコーン系溶剤が例示される。溶剤を使用する場合、外気への汚染の問題から、溶剤の沸点は、150℃以上が好ましく、200℃以上がより好ましく、250℃以上が特に好ましい。これらの溶剤は、単独で使用してもよく、2種以上併用してもよい。
但し、溶剤の配合量が多い場合には、環境への影響や人体への毒性が高くなる場合があるため、溶剤の使用量を低減する事が好ましい。その為、溶剤の配合量は、(A)成分と(B)成分の総量100質量部に対して、50質量部以下であることが好ましく、30質量部以下であることがより好ましく、10質量部以下であることが更に好ましく、溶剤を含まないことが特に好ましい。
本発明の硬化性樹脂組成物には、必要に応じて粘着性付与剤を添加することができる。粘着性付与剤としては、特に限定されないが、常温で固体、液体を問わず通常使用されるものを使用することができる。具体例としては、スチレン系ブロック共重合体、その水素添加物、フェノール樹脂、変性フェノール樹脂(例えば、カシューオイル変性フェノール樹脂、トール油変性フェノール樹脂等)、テルペンフェノール樹脂、キシレン-フェノール樹脂、シクロペンタジエン-フェノール樹脂、クマロンインデン樹脂、ロジン系樹脂、ロジンエステル樹脂、水添ロジンエステル樹脂、キシレン樹脂、低分子量ポリスチレン系樹脂、スチレン共重合体樹脂、石油樹脂(例えば、C5炭化水素樹脂、C9炭化水素樹脂、C5C9炭化水素共重合樹脂等)、水添石油樹脂、テルペン系樹脂、DCPD樹脂石油樹脂等が挙げられる。これらは単独で用いても良く、2種以上を併用しても良い。スチレン系ブロック共重合体及びその水素添加物としては、スチレン-ブタジエン-スチレンブロック共重合体(SBS)、スチレン-イソプレン-スチレンブロック共重合体(SIS)、スチレン-エチレンブチレン-スチレンブロック共重合体(SEBS)、スチレン-エチレンプロピレン-スチレンブロック共重合体(SEPS)、スチレン-イソブチレン-スチレンブロック共重合体(SIBS)等が挙げられる。上記粘着性付与剤は単独で用いてもよく、2種以上併用してもよい。
本発明の組成物には、必要に応じてレベリング剤を添加することができる。レベリング剤としては市販されているものを使用することができる。市販品としては、例えば、BYKETOL(登録商標)-OK、BYKETOL(登録商標)-SPECIAL、BYKETOL(登録商標)-AQ、BYKETOL(登録商標)-WS(いずれもビックケミー社製)、DISPARLON(登録商標) 1970、DISPARLON(登録商標) 230、DISPARLON(登録商標) LF-1980、DISPARLON(登録商標) LF-1982、DISPARLON(登録商標) LF-1983、DISPARLON(登録商標) LF-1984、DISPARLON(登録商標) LF-1985(いずれも楠本化成社製)等を挙げることができる。
本発明の硬化性樹脂組成物には、必要に応じて垂れを防止し、作業性を良くするためにチクソ性付与剤(垂れ防止剤)を添加しても良い。垂れ防止剤としては特に限定されないが、例えば、ポリアミドワックス類;水添ヒマシ油誘導体類;ステアリン酸カルシウム、ステアリン酸アルミニウム、ステアリン酸バリウム等の金属石鹸類等が挙げられる。充填材として示した前記ヒュームドシリカもまた、チクソ性付与剤として使用できる。また、特開平11-349916号公報に記載されているような粒子径10~500μmのゴム粉末や、特開2003-155389号公報に記載されているような有機質繊維を用いると、チクソ性が高く作業性の良好な組成物が得られる。これらチクソ性付与剤(垂れ防止剤)は単独で用いてもよく、2種以上併用してもよい。
本発明の硬化性樹脂組成物には、必要に応じてエポキシ樹脂を添加しても良い。
エポキシ樹脂としては、例えば、ビスフェノールA型エポキシ樹脂、ビスフェノールF型エポキシ樹脂、ノボラック型エポキシ樹脂、グリシジルエステル型エポキシ樹脂、水添ビスフェノールA(又はF)型エポキシ樹脂、グリシジルエーテル型エポキシ樹脂、含アミノグリシジルエーテル樹脂やこれらのエポキシ樹脂に、ビスフェノールA(又はF)類、多塩基酸類等を付加反応させて得られるエポキシ化合物等、公知のエポキシ樹脂が挙げられる。
本発明の硬化性樹脂組成物には、必要に応じて酸化防止剤(老化防止剤)を使用することができる。酸化防止剤を使用すると硬化物の耐熱性を高めることができる。酸化防止剤としてはヒンダードフェノール系、モノフェノール系、ビスフェノール系、ポリフェノール系が例示できるが、特にヒンダードフェノール系が好ましい。同様に、チヌビン(登録商標)622LD,チヌビン(登録商標)144,CHIMASSORB(登録商標)944LD,CHIMASSORB(登録商標)119FL(以上いずれもチバ・スペシャルティ・ケミカルズ株式会社製);MARK LA-57,MARK LA-62,MARK LA-67,MARK LA-63,MARK LA-68(以上いずれも旭電化工業株式会社製);サノール(登録商標)LS-770,サノール(登録商標)LS-765,サノール(登録商標)LS-292,サノール(登録商標)LS-2626,サノール(登録商標)LS-1114,サノール(登録商標)LS-744(以上いずれも三共株式会社製)に示されたヒンダードアミン系光安定剤を使用することもできる。
本発明の硬化性樹脂組成物には、必要に応じて光安定剤を使用することができる。光安定剤を使用すると硬化物の光酸化劣化を防止できる。光安定剤としてベンゾトリアゾール系、ヒンダードアミン系、ベンゾエート系化合物等が例示できるが、よりヒンダードアミン系化合物が好ましい。特に3級アミン含有ヒンダードアミン系光安定剤を用いるのが組成物の保存安定性改良のために好ましい。3級アミン含有ヒンダードアミン系光安定剤としてはチヌビン(登録商標)622LD,チヌビン(登録商標)144,CHIMASSORB(登録商標)119FL(以上いずれもBASF製);MARK LA-57,LA-62,LA-67,LA-63(以上いずれも株式会社アデカ製);サノール(登録商標)LS-765,LS-292,LS-2626,LS-1114,LS-744(以上いずれも三共株式会社製)などの光安定剤が例示できる。
本発明の硬化性樹脂組成物には、必要に応じて紫外線吸収剤を使用することができる。紫外線吸収剤を使用すると硬化物の表面耐候性を高めることができる。紫外線吸収剤としてはベンゾフェノン系、ベンゾトリアゾール系、サリシレート系、置換トリル系及び金属キレート系化合物等が例示できるが、特にベンゾトリアゾール系化合物が好ましい。
本発明の硬化性樹脂組成物には、必要に応じてシランカップリング剤を添加することができる。シランカップリング剤添加により接着性を向上させることができる。具体例としては、γ-イソシアネートプロピルトリメトキシシラン、γ-イソシアネートプロピルトリエトキシシラン、γ-イソシアネートプロピルメチルジエトキシシラン、γ-イソシアネートプロピルメチルジメトキシシラン、(イソシアネートメチル)トリメトキシシラン、(イソシアネートメチル)ジメトキシメチルシラン、(イソシアネートメチル)トリエトキシシラン、(イソシアネートメチル)ジエトキシメチルシラン等のイソシアネート基含有シラン類;γ-アミノプロピルトリメトキシシラン、γ-アミノプロピルトリエトキシシラン、γ-アミノプロピルトリイソプロポキシシラン、γ-アミノプロピルメチルジメトキシシラン、γ-アミノプロピルメチルジエトキシシラン、γ-(2-アミノエチル)アミノプロピルトリメトキシシラン、γ-(2-アミノエチル)アミノプロピルメチルジメトキシシラン、γ-(2-アミノエチル)アミノプロピルトリエトキシシラン、γ-(2-アミノエチル)アミノプロピルメチルジエトキシシラン、γ-(2-アミノエチル)アミノプロピルトリイソプロポキシシラン、γ-(6-アミノヘキシル)アミノプロピルトリメトキシシラン、3-(N-エチルアミノ)-2-メチルプロピルトリメトキシシラン、γ-ウレイドプロピルトリメトキシシラン、γ-ウレイドプロピルトリエトキシシラン、N-フェニル-γ-アミノプロピルトリメトキシシラン、N-ベンジル-γ-アミノプロピルトリメトキシシラン、N-ビニルベンジル-γ-アミノプロピルトリエトキシシラン、N-シクロヘキシルアミノメチルトリエトキシシラン、N-シクロヘキシルアミノメチルジエトキシメチルシラン、N-フェニルアミノメチルトリメトキシシラン、(2-アミノエチル)アミノメチルトリメトキシシラン、N,N’-ビス[3-(トリメトキシシリル)プロピル]エチレンジアミン等のアミノ基含有シラン類;N-(1,3-ジメチルブチリデン)-3-(トリエトキシシリル)-1-プロパンアミン等のケチミン型シラン類;γ-メルカプトプロピルトリメトキシシラン、γ-メルカプトプロピルトリエトキシシラン、γ-メルカプトプロピルメチルジメトキシシラン、γ-メルカプトプロピルメチルジエトキシシラン、メルカプトメチルトリメトキシシラン、メルカプトメチルトリエトキシシラン等のメルカプト基含有シラン類;γ-グリシドキシプロピルトリメトキシシラン、γ-グリシドキシプロピルトリエトキシシラン、γ-グリシドキシプロピルメチルジメトキシシラン、β-(3,4-エポキシシクロヘキシル)エチルトリメトキシシラン、β-(3,4-エポキシシクロヘキシル)エチルトリエトキシシラン等のエポキシ基含有シラン類;β-カルボキシエチルトリエトキシシラン、β-カルボキシエチルフェニルビス(2-メトキシエトキシ)シラン、N-β-(カルボキシメチル)アミノエチル-γ-アミノプロピルトリメトキシシラン等のカルボキシシラン類;ビニルトリメトキシシラン、ビニルトリエトキシシラン、γ-メタクリロイルオキシプロピルメチルジメトキシシラン、γ-アクリロイルオキシプロピルトリエトキシシラン、メタクリロイルオキシメチルトリメトキシシラン等のビニル型不飽和基含有シラン類;γ-クロロプロピルトリメトキシシラン等のハロゲン含有シラン類;トリス(3-トリメトキシシリルプロピル)イソシアヌレート等のイソシアヌレートシラン類等を挙げることができる。また、これらを変性した誘導体である、アミノ変性シリルポリマー、シリル化アミノポリマー、不飽和アミノシラン錯体、フェニルアミノ長鎖アルキルシラン、アミノシリル化シリコーン、シリル化ポリエステル等もシランカップリング剤として用いることができる。シランカップリング剤の反応物としては、アミノシランとエポキシシランの反応物、アミノシランとイソシアネートシランの反応物、各種シランカップリング剤の部分縮合体等を挙げる事ができる。
本発明の硬化性樹脂組成物には、必要に応じて(E)成分として脱水剤を添加することができる。脱水剤添加により組成物中に存在する水分を除去でき、保存安定性や硬化時の発泡などが改善される。具体例としては、ビニルトリメトキシシラン、酸化カルシウム、ゼオライト、p-トルエンスルホニルイソシアネート、3-エチル-2-メチル-2-(3-メチルブチル)-1,3-オキサゾリジン等のオキサゾリジン類などが挙げられ、これらは単独あるいは2種以上を組合わせて使用できる。
本発明では、必要に応じて、その他の配合成分を使用することができる。その他の配合成分としては、加水分解安定剤、チタネート系カップリング剤、アルミネート系カップリング剤、離型剤、帯電防止剤、滑剤、低収縮剤、シリコーン界面活性剤等が挙げられる。
本発明の硬化性樹脂組成物は、(A)成分および(B)成分を主成分とする硬化性樹脂組成物中に、ポリマー微粒子(C)を含有する組成物であり、好ましくは、ポリマー微粒子(C)が1次粒子の状態で分散した組成物である。
本発明の硬化性樹脂組成物を2液型または多液型として使用する場合、本発明の各成分は、イソシアネート基含有成分<(B)成分、および/または、1より大きい当量比(NCO/活性水素含有基)で(A)成分と(B)成分を反応させて得たイソシアネート基を有するウレタンプレポリマー)>を含有する第1液と、水酸基を有する成分<(A)成分、および/または、1未満の当量比(NCO/活性水素含有基)で(A)成分と(B)成分を反応させて得た水酸基を有するウレタンプレポリマー>を含有する第2液として、別々の容器に保管し、使用直前に混合して使用する事が好ましい。この際、(A)成分、(B)成分、及び、前記ウレタンプレポリマー以外の成分、すなわち、(C)成分、(D)成分、及び、その他の配合成分は、前記の第1液に添加してもよく、前記の第2液に添加してもよい。保存安定性の点から、前記(B)成分を含有する第1液と、前記(A)成分と前記(C)成分と前記(D)成分を含有する第2液とからなる2液型硬化性樹脂組成物とする態様も本発明に含まれる。
本発明には、上記硬化性樹脂組成物を硬化して得られる硬化物が含まれる。本発明の硬化性樹脂組成物は、ポリマー微粒子が一次粒子の状態で分散していることから、これを硬化することによって、ポリマー微粒子が均一に分散した硬化物を容易に得ることができる。また、ポリマー微粒子が膨潤し難く、硬化性樹脂組成物の粘性が低いことから、硬化物を作業性よく得ることができる。
この場合、3官能以上のポリエーテルポリオールの量は、ポリオール100質量%中、20質量%以上であることが好ましく、より好ましくは30質量%以上、さらに好ましくは40質量%以上、さらにより好ましくは50質量%以上であり、100質量%以下であることが好ましく、より好ましくは99質量%以下、さらに好ましくは98質量%以下、さらにより好ましくは97質量%以下である。当該量が20質量%未満の場合、ガラス転移温度の向上が見込めない虞がある。
4官能のポリエーテルポリオールの量は、ポリオール100質量%中、50質量%以上であることが好ましく、より好ましくは55質量%以上、さらに好ましくは60質量%以上であり、90質量%以下であることが好ましく、より好ましくは85質量%以下、さらに好ましくは80質量%以下である。当該量が50質量%未満の場合、ガラス転移温度の向上が見込めない虞があり、当該量が90質量%超である場合、剛性が高くなりすぎて、靭性が低下する虞がある。
本発明の硬化性樹脂組成物は、構造接着、インキバインダー、木材チップバインダー、ゴムチップ用バインダー、フォームチップバインダー、鋳物用バインダー、床材、セラミック、岩盤固結材、自動車内装材、一般木工、家具、内装、壁材、食品包装等の接着剤;コーティング材;繊維強化複合材料;自動車シート、自動車内装部品、吸音材、制振材、ショックアブソーバー、断熱材、工事用床材クッション等のウレタンフォーム;等の用途に好ましく用いられる。
本発明の硬化性樹脂組成物は、冷間圧延鋼、アルミニウム、ファイバーグラスで強化されたポリエステル(FRP)、炭素繊維で強化されたエポキシ樹脂等の熱硬化性樹脂の硬化物のパネル、炭素繊維で強化された熱可塑性樹脂シートのパネル、シートモウルディングコンパウンド(SMC)、ABS、PVC、ポリカーボネート、ポリプロピレン、TPO、木材およびガラス等、種々の被着体へ良好な接着性を示す。
本発明の硬化性樹脂組成物をコーティング材として使用する場合、一般にコテやレーキを使用する場合は、混合物の粘度は500~9,000cps/25℃程度とし、またローラーやスプレーの場合は、100~3,000cps/25℃程度に調製する。本発明の硬化性樹脂組成物を、例えば床や廊下に施工するには、一般に実施されているウレタン塗り床材の施工法が適用できる。たとえば、素地調整した下地にプライマーを塗布後、施工条件に応じて、コテ、ローラー、レーキ、スプレーガンなどを用いて均一に塗工する。塗工後、硬化が進み、性能の良いウレタン舗装膜が得られる。本発明の硬化性樹脂組成物を硬化して得た塗膜は、耐荷重性や耐摩耗性に優れた塗膜が得られる。
本発明の硬化性樹脂組成物を繊維強化複合材料として使用する場合、強化繊維としては、特に限定されないが、ガラス繊維、ガラス長繊維、炭素繊維、天然繊維、金属繊維、熱可塑性樹脂繊維、ボロン繊維、アラミド繊維、ポリエチレン繊維、ザイロン強化繊維等が挙げられる。特に、ガラス繊維や炭素繊維が好ましい。
本発明の硬化性樹脂組成物を発泡してなる発泡体も本発明に含まれる態様である。
前記発泡体は、例えば、上記の通り、ポリマー微粒子(C)を分散させてなるポリオール(A)と、ポリイソシアネート(B)とを、硬化触媒、発泡剤および整泡剤の存在下で反応させる方法で製造されてもよい。
発泡剤は、特に限定されるものではなく、ジクロロモノフルオロエタン、ハイドロフルオロカーボンなどのフロン系発泡剤を用いることもできるが、環境汚染の点から、水を用いることが好ましい。水はポリイソシアネートとの反応により生成する炭酸ガスを利用するために、環境上の問題がない。
発泡剤の配合量は、ポリオール(A)成分100質量部に対して、通常0.01~10質量部の範囲であり、0.1~5質量部の範囲が好ましい。
整泡剤は、通常用いられるものであれば、特に限定されるものではなく、例えば、各種シロキサン-ポリエーテルブロック共重合体等のシリコーン系界面活性剤を用いることができる。
整泡剤の配合量は、ポリオール(A)成分100質量部に対して、通常0.01~5質量部であり、0.1~3質量部とすることが好ましい。
先ず、実施例および比較例によって製造した硬化性樹脂組成物の評価方法について、以下説明する。
水性ラテックスに分散しているポリマー微粒子の体積平均粒子径(Mv)は、マイクロトラックUPA150(日機装株式会社製)を用いて測定した。脱イオン水で希釈したものを測定試料として用いた。測定は、水の屈折率、およびそれぞれのポリマー微粒子の屈折率を入力し、計測時間600秒、Signal Levelが0.6~0.8の範囲内になるように試料濃度を調整して行った。
硬化板サンプルを、長さ100mm、幅(b)10mm、厚さ(h)5mmのサイズの試験片に切削後、23℃で養生、その後、オートグラフAG-2000E(島津製作所製)を用いて、支点間距離(L)80mm、テストスピード2mm/分の条件にて3点曲げ試験を実施した。得られた荷重(F)-たわみ(e)曲線の初期傾き(F/e)を求め、曲げ弾性率(E)を下記の数式1より算出した。また、最大曲げ応力時のたわみ(efM)から、最大曲げ応力時の曲げひずみ(εfM)を下記の数式2より算出した。ここで、(F/e)はkN/mm単位、L、b、h、efMはmm単位である。
E(GPa)=L3×(F/e)/(4×b×h3) <数式1>
εfM(%)=600×efM×h/L2 <数式2>
硬化板サンプルを長さ2.5インチ、幅(b)0.5インチ、厚さ(h)5mmのサイズの試験片に切削後、ノッチングマシーンによりVノッチを入れた。その後、Vノッチ先端からカミソリ刃を用いて試験片中央までクラックを入れた。試験片を23℃で養生後、オートグラフAG-2000E(島津製作所製)を用い、支点間距離(L)50mm、テストスピード1mm/分の条件で3点曲げ試験を行なった。曲げ試験から得られた最大強度F(kN)を用い、下記の数式2、及び数式3に従い、破壊靱性値K1c(MPa・m1/2)を算出した。ここで、aはVノッチの深さとVノッチ先端からクラック先端までの長さの和であり、L、h、a、及びbはcm単位である。
K1c=(F×L/(h×b3/2))×f <数式2>
f=3(a/b)1/2×AA/BB <数式3>
AA=1.99-(a/b)×{1-(a/b)}
×{2.15-3.93(a/b)+2.7(a/b)2}
BB=2{1+2(a/b)}{1-(a/b)}3/2
JIS K 6854に従って、寸法:25×200×0.5mmの2枚の冷間圧延鋼板(SPCC-SD)の間に、硬化性樹脂組成物を塗布し、貼り合せて、接着剤厚みを250μmとした。これを、80℃で3時間硬化させた。その後、試験片を23℃で、オートグラフAG-2000E(島津製作所製)を用い、テストスピード254mm/分の条件で180°剥離試験を行なった。
硬化性樹脂組成物を、寸法:20×90×0.8mmの2枚の冷間圧延鋼板(SPCC-SD)の間に塗布し、接着層厚み0.25mmとなるように重ね合せ、80℃で3時間硬化させ、ISO 11343に従って、23℃での動的割裂抵抗力を測定した。
製造例1-1;ポリブタジエンゴムラテックス(R-1)の調製
100L耐圧重合機中に、脱イオン水200質量部、リン酸三カリウム0.03質量部、リン酸二水素カリウム0.25質量部、エチレンジアミン四酢酸二ナトリウム(EDTA)0.002質量部、硫酸第一鉄・7水和塩(Fe)0.001質量部およびドデシルベンゼンスルホン酸ナトリウム(SDS)1.5質量部を投入し、撹拌しつつ十分に窒素置換を行なって酸素を除いた後、ブタジエン(BD)100質量部を系中に投入し、45℃に昇温した。パラメンタンハイドロパーオキサイド(PHP)0.015質量部、続いてナトリウムホルムアルデヒドスルホキシレート(SFS)0.04質量部を投入し重合を開始した。重合開始から4時間目に、PHP0.01質量部、EDTA0.0015質量部およびFe0.001質量部を投入した。重合10時間目に減圧下残存モノマーを脱揮除去して重合を終了し、ポリブタジエンゴム粒子を含むラテックス(R-1)を得た。得られたラテックスに含まれるポリブタジエンゴム粒子の体積平均粒子径は0.10μmであった。
耐圧重合機中に、製造例1-1で得たポリブタジエンゴムラテックス(R-1)を21質量部(ポリブタジエンゴム7質量部を含む)、脱イオン水185質量部、リン酸三カリウム0.03質量部、EDTA0.002質量部、及び硫酸第一鉄・7水和塩0.001質量部を投入し、撹拌しつつ十分に窒素置換を行なって酸素を除いた後、ブタジエン(BD)93質量部を系中に投入し、45℃に昇温した。PHP0.02質量部、続いてSFS0.10質量部を投入し重合を開始した。重合開始から24時間目まで3時間おきに、それぞれ、PHP0.025質量部、及びEDTA0.0006質量部、及び硫酸第一鉄・7水和塩0.003質量部を投入した。重合30時間目に減圧下残存モノマーを脱揮除去して重合を終了し、ポリブタジエンゴムを主成分とするポリブタジエンゴムラテックス(R-2)を得た。得られたラテックスに含まれるポリブタジエンゴム粒子の体積平均粒子径は0.20μmであった。
製造例2-1;コアシェルポリマーラテックス(L-1)の調製
3Lガラス容器に、製造例1-1で得たラテックス(R-1)1575質量部(ポリブタジエンゴム粒子510質量部相当)および脱イオン水315質量部を仕込み、窒素置換を行いながら60℃で撹拌した。EDTA0.024質量部、Fe0.006質量部、SFS1.2質量部を加えた後、グラフトモノマー(メチルメタクリレート(MMA)60質量部、4-ヒドロキシブチルアクリレート(4HBA)30質量部)、およびクメンヒドロパーオキサイド(CHP)0.3質量部の混合物を2時間かけて連続的に添加しグラフト重合した。添加終了後、更に2時間撹拌して反応を終了させ、コアシェルポリマー(C-1)のラテックス(L-1)を得た。得られたラテックスに含まれるコアシェルポリマーの体積平均粒子径は0.11μmであった。
温度計、撹拌機、還流冷却器、窒素流入口、及びモノマーの添加装置を有するガラス反応器に、製造例1-2で得たポリブタジエンゴムラテックス(R-2)240質量部(ポリブタジエンゴム粒子80質量部を含む)、及び脱イオン水57質量部を仕込み、窒素置換を行いながら60℃で撹拌した。EDTA0.004質量部、硫酸第一鉄・7水和塩0.001質量部、及びSFS0.2質量部を加えた後、MMA18質量部、スチレン(ST)2質量部、及び、CHP0.06質量部の混合物を85分間かけて連続的に添加した。添加終了後、CHP0.04質量部を添加し、さらに1時間撹拌を続けて重合を完結させ、コアシェルポリマー(C-2)を含む水性ラテックス(L-2)を得た。モノマー成分の重合転化率は99%以上であった。得られた水性ラテックスに含まれるコアシェルポリマーの体積平均粒子径は0.21μmであった。
製造例2-2において、グラフトモノマーとして<MMA18質量部、スチレン(ST)2質量部>の代わりに<MMA17.5質量部、ST2質量部、4HBA0.5質量部>を用いたこと以外は製造例2-2と同様にして、コアシェルポリマー(C-3)のラテックス(L-3)を得た。得られたラテックスに含まれるコアシェルポリマーの体積平均粒子径は0.21μmであった。
製造例2-2において、グラフトモノマーとして<MMA18質量部、スチレン(ST)2質量部>の代わりに<MMA17質量部、ST2質量部、4HBA1質量部>を用いたこと以外は製造例2-2と同様にして、コアシェルポリマー(C-4)のラテックス(L-4)を得た。得られたラテックスに含まれるコアシェルポリマーの体積平均粒子径は0.21μmであった。
製造例2-2において、グラフトモノマーとして<MMA18質量部、スチレン(ST)2質量部>の代わりに<MMA16質量部、ST2質量部、4HBA2質量部>を用いたこと以外は製造例2-2と同様にして、コアシェルポリマー(C-5)のラテックス(L-5)を得た。得られたラテックスに含まれるコアシェルポリマーの体積平均粒子径は0.21μmであった。
製造例2-2において、グラフトモノマーとして<MMA18質量部、スチレン(ST)2質量部>の代わりに<MMA14質量部、ST2質量部、4HBA4質量部>を用いたこと以外は製造例2-2と同様にして、コアシェルポリマー(C-6)のラテックス(L-6)を得た。得られたラテックスに含まれるコアシェルポリマーの体積平均粒子径は0.21μmであった。
製造例3-1;分散物(M-1)の調製
25℃の1L混合槽にメチルエチルケトン(MEK)132gを導入し、撹拌しながら、それぞれ前記製造例2-1で得られたコアシェルポリマー(C-1)の水性ラテックス(L-1)を132g(ポリマー微粒子40g相当)投入した。均一に混合後、水200gを80g/分の供給速度で投入した。供給終了後、速やかに撹拌を停止したところ、浮上性の凝集体および有機溶媒を一部含む水相からなるスラリー液を得た。次に、一部の水相を含む凝集体を残し、水相360gを槽下部の払い出し口より排出させた。得られた凝集体にMEK90gを追加して均一に混合し、コアシェルポリマーを均一に分散した分散体を得た。この分散体に、(A)成分である3官能ポリエーテルポリオールのPPT300(A-1:三井化学製、アクトコール T-300)80gを混合した。この混合物から、回転式の蒸発装置で、MEKを除去した。このようにして、(A)成分にポリマー微粒子が分散した分散物(M-1)を得た。
製造例3-1において、PPT300(A-1)の代わりに2官能ポリエーテルポリオールのPPG1000(A-2:三井化学製、アクトコール D-1000)を用いたこと以外は製造例3-1と同様にして、ポリマー微粒子が分散した分散物(M-2)を得た。
製造例3-1において、PPT300(A-1)の代わりに2官能ポリエーテルポリオールのPPG400(A-3:三井化学製、アクトコール D-400)を用いたこと以外は製造例3-1と同様にして、ポリマー微粒子が分散した分散物(M-3)を得た。
25℃の1L混合槽にメチルエチルケトン(MEK)132gを導入し、撹拌しながら、それぞれ前記製造例2-2~2-6で得られたコアシェルポリマー(C-2~C-6)の水性ラテックス(L-2~L-6)を132g(ポリマー微粒子40g相当)投入した。均一に混合後、水200gを80g/分の供給速度で投入した。供給終了後、速やかに撹拌を停止したところ、浮上性の凝集体および有機溶媒を一部含む水相からなるスラリー液を得た。次に、一部の水相を含む凝集体を残し、水相360gを槽下部の払い出し口より排出させた。得られた凝集体にMEK90gを追加して均一に混合し、コアシェルポリマーを均一に分散した分散体を得た。この分散体に、(A)成分であるポリエステルポリオール(A-6:Bayer社製、Desmophen 1200、水酸基含有量5.0%)80gを混合した。この混合物から、回転式の蒸発装置で、MEKを除去した。このようにして、(A)成分にポリマー微粒子が分散した分散物(M-4~M-8)を得た。
表1に示す処方にしたがって、(A)成分であるPPT300(A-1)またはPPG1000(A-2)、前記製造例3-1~2で得られた分散物(M-1~2)、(B)成分であるイソホロンジイソシアネート(B-1:和光純薬工業製)、(D)成分であるDBUのオクチル酸塩(サンアプロ製:U-CAT SA102)、脱水剤として、粉末状の合成ゼオライトA-3(200mesh通過分:和光純薬工業製)をそれぞれ計量し、よく混合して脱泡し硬化性樹脂組成物を得た。なお、表1の組成物中の(B)成分のイソシアネート基の総モル量(β)と(A)成分の水酸基の総モル量(α)との比の値(β/α)は、全て1.0である。表1の各組成物を、厚み5mmのスペーサーを挟んだ2枚のテフロン(登録商標)コート鋼板の間に注ぎ込み、熱風オーブン中80℃で3時間硬化させ、厚み5mmの硬化板を得た。実施例1~2と比較例1~2の硬化板は硬質の硬化板であった為、この硬化板を用いて前記の試験方法に従って、最大曲げ応力時の曲げひずみと破壊靭性K1cを測定した。試験結果を表1に示す。なお、実施例2と比較例2では、破壊靭性の測定の際に不安定破壊が起きず、有効なK1cの値が得られなかった。一方、比較例3~4の硬化板は軟質の硬化板であった為、曲げ物性や破壊靱性の測定ができなかった。そこで、JIS K 6251に従って、前記硬化板から3号ダンベル試験片を採取し、500mm/minの速度で23℃で引張試験を行い、その際の最大引張応力時の伸びを測定した。試験結果を表1に示す。

表2に示す処方にしたがって、(A)成分である3官能ポリエーテルポリオールのPPT300(A-1:三井化学製、アクトコール T-300)、2官能ポリエーテルポリオールのPPG400(A-3:三井化学製、アクトコール D-400)、2官能ポリエーテルポリオールのPPG200(A-4:三洋化成製、サンニックスPP-200)、4官能ポリエーテルポリオールPPQ(A-5:三洋化成製、サンニックスHD―402)、前記製造例3-1または製造例3-3で得られた分散物(M-1またはM-3)を混合して混合液を作製した。この混合液を、60℃に加温保持しながら、乾燥窒素を溶液に1時間吹き込む、あるいは脱水剤(粉末状の合成ゼオライトA-3(200mesh通過分:和光純薬工業製)を添加することで混合液を乾燥した。なお、脱水剤を使用した場合は、次の工程では、脱水剤を除かず、そのまま使用した。つづいて、得られた混合液を、(B)成分であるポリメリックMDI(B-2:三井化学製、コスモネートM-200)および、必要あれば消泡剤(ビックケミー製、BYK(登録商標)-A500)とを、よく混合して脱泡し硬化性樹脂組成物を得た。なお、表2の組成物中の(B)成分のイソシアネート基の総モル量(β)と(A)成分の水酸基の総モル量(α)との比の値(β/α)は、全て1.1である。この組成物を、厚み5mmのスペーサーを挟んだ2枚のテフロン(登録商標)コート鋼板の間に注ぎ込み、熱風オーブン中130℃で3時間硬化させ、厚み5mmの硬化板を得た。この硬化板を用いて前記の試験方法に従って、曲げ弾性率および破壊靭性K1cを測定した。また、この硬化板のガラス転移温度をDSC(示差走査熱量測定)により測定した。試験結果を表2に示す。
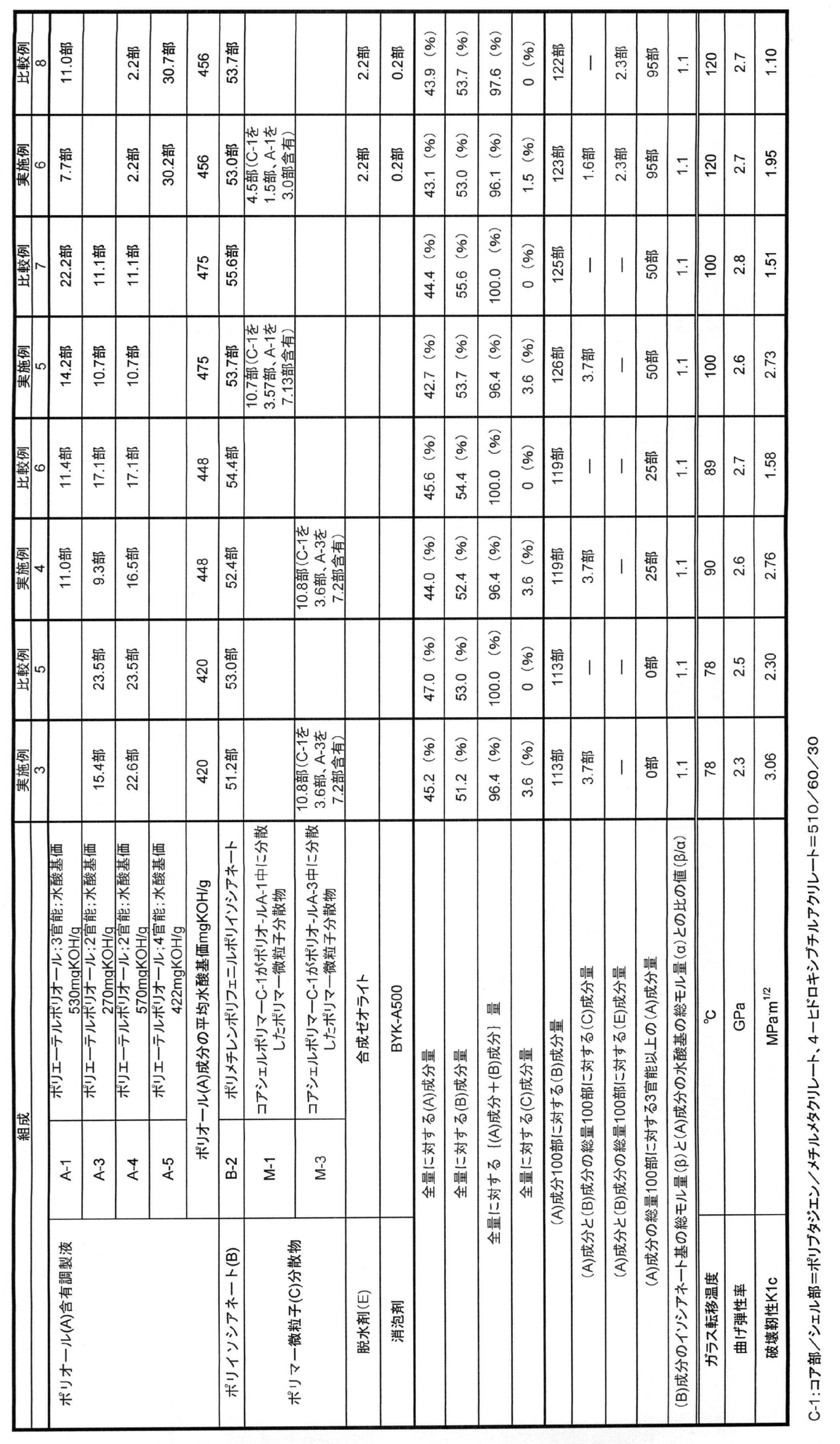
表3に示す処方にしたがって、(A)成分である3官能ポリエーテルポリオールのPPT300(A-1:三井化学製、アクトコール T-300)、2官能ポリエーテルポリオールのPPG400(A-3:三井化学製、アクトコール D-400)、2官能ポリエーテルポリオールのPPG200(A-4:三洋化成製、サンニックスPP-200)、前記製造例3-1で得られた分散物(M-1)を混合して混合液を作製した。この混合液に脱水剤(粉末状の合成ゼオライトA-3(200mesh通過分:和光純薬工業製)、および充填剤であるシリカ(龍森製、クリスタライトC)を添加してよく混合した。なお、脱水剤を使用した場合は、次の工程では、脱水剤を除かず、そのまま使用した。つづいて、ポリオール・シリカ混合物を、(B)成分であるポリメリックMDI(B-2:三井化学製、コスモネートM-200)と、よく混合して脱泡し硬化性樹脂組成物を得た。なお、表3の組成物中の(B)成分のイソシアネート基の総モル量(β)と(A)成分の水酸基の総モル量(α)との比の値(β/α)は、全て1.1である。この組成物を、厚み5mmのスペーサーを挟んだ2枚のテフロン(登録商標)コート鋼板の間に注ぎ込み、熱風オーブン中110℃で3時間硬化させ、厚み5mmの硬化板を得た。この硬化板を用いて前記の試験方法に従って、ガラス転移温度、曲げ弾性率および破壊靭性K1cを測定した。試験結果を表3に示す。

表4に示す処方にしたがって、(A)成分である3官能ポリエーテルポリオールのPPT300(A-1:三井化学製、アクトコール T-300)、2官能ポリエーテルポリオールのPPG400(A-3:三井化学製、アクトコール D-400)、2官能ポリエーテルポリオールのPPG200(A-4:三洋化成製、サンニックスPP-200)、前記製造例3-1で得られた分散物(M-1)を混合して混合液を作製した。この混合液を、60℃に加温保持しながら、乾燥窒素を混合液に1時間吹き込んで乾燥した。つづいて、得られた混合液に、発泡剤である水、整泡剤であるシリコーン系界面活性剤(東レダウコーニング社製、SH193)、(B)成分であるポリメリックMDI(B-2:三井化学製、コスモネートM-200)、(D)成分である1,4-ジアザビシクロ[2,3,2]オクタン溶液(シグマ・アルドリッチ製、DABCO 33-LV)とを加えて、よく混合し硬化性樹脂組成物を得た。なお、表4の組成物中の(B)成分のイソシアネート基の総モル量(β)と(A)成分の水酸基の総モル量(α)との比の値(β/α)は、全て1.1である。ついで、この組成物を、40℃に温調したテフロン(登録商標)コート鋼板からなる成形型に注ぎ込み、厚み5mmの発泡成形体を得た。この発泡成形体の重量と見かけ体積と未発泡成形体の比重から、成形密度を測定した。また、JIS K7111-1に従って、この発泡成形体を用いてシャルピー強度を測定した。試験結果を表4に示す。

表5に示す処方にしたがって、(A)成分である3官能ポリエーテルポリオールのPPT300(A-1:三井化学製、アクトコール T-300)、2官能ポリエーテルポリオールのPPG400(A-3:三井化学製、アクトコール D-400)、2官能ポリエーテルポリオールのPPG200(A-4:三洋化成製、サンニックスPP-200)、前記製造例3-1で得られた分散物(M-1)を混合して混合液を作製した。この混合液を、60℃に加温保持しながら、乾燥窒素を混合液に1時間吹き込むことで混合液を乾燥した。つづいて、得られた混合液を、(B)成分であるポリメリックMDI(B-2:三井化学製、コスモネートM-200)を、よく混合して脱泡し硬化性樹脂組成物を得た。なお、表5の組成物中の(B)成分のイソシアネート基の総モル量(β)と(A)成分の水酸基の総モル量(α)との比の値(β/α)は、全て1.1である。この組成物を、195g/m2の炭素繊維織物を8枚重ねた型に、吸引したのち、100℃で3時間加熱することで、炭素繊維強化成形体を得た。この得られた成形体を、長さ50mm、幅5mmのサイズの試験片に切削後、支点間距離を30mmに設定した3点曲げ試験を実施し、試験片が破壊するまでの変形量を比較した。変形量が大きい方が割れにくく、優れている。相対的により変形量が大きいものを「優」、小さいものを「劣」とした。結果を表5に示す。

表6に示す処方にしたがって、(A)成分であるPPT300(A-1)またはPPG1000(A-2)、前記製造例3-1~2で得られた分散物(M-1~2)、(B)成分であるポリメリックMDI(B-2:三井化学製、コスモネートM-200)、(D)成分であるDBUのオクチル酸塩(サンアプロ製:U-CAT SA102)、膠質炭酸カルシウム(白石工業製、白艶華CCR)、脱水剤として、粉末状の合成ゼオライトA-3(200mesh通過分:和光純薬工業製)をそれぞれ計量し、よく混合して脱泡し硬化性樹脂組成物を得た。なお、表6の組成物中の(B)成分のイソシアネート基の総モル量(β)と(A)成分の水酸基の総モル量(α)との比の値(β/α)は、全て1.1である。この組成物を用いて前記の試験方法に従って、T字剥離強度を測定した。試験結果を表6に示す。

表7に示す処方にしたがって、(A)成分であるポリエステルポリオール(A-6)またはポリエーテルポリオールPPG1000(A-2)、前記製造例3-2および製造例3-4~8で得られた分散物(M-2およびM-4~8)、(B)成分であるポリメリックMDI(B-2:三井化学製、コスモネートM-200)、ヒュームドシリカ(CABOT製、CAB-O-SIL TS-720)、脱水剤として、粉末状の合成ゼオライトA-3(200mesh通過分:和光純薬工業製)をそれぞれ計量し、よく混合して脱泡し硬化性樹脂組成物を得た。なお、表7の組成物中の(B)成分のイソシアネート基の総モル量(β)と(A)成分の水酸基の総モル量(α)との比の値(β/α)は、全て1.1である。この組成物を用いて前記の試験方法に従って、T字剥離強度と動的割裂抵抗力(耐衝撃剥離接着性)を測定した。試験結果を表7に示す。

実施例1の硬化性樹脂組成物を用いて、モルタル板上に100μmの厚みで塗布した。これを80℃で3時間硬化し、積層体を得た。得られた積層体の塗膜は、衝撃に強い塗膜であった。この積層体の塗膜を室外の日当たりのよい場所に1年間放置したところ、塗膜の黄変が観察された。
実施例1の硬化性樹脂組成物の、(D)成分であるDBUのオクチル酸塩(サンアプロ製:U-CAT SA102)1質量部を、ジブチル錫ジラウレート0.01質量部に変更した硬化性樹脂組成物を用いて、実施例18と同様に、モルタル板上に100μmの厚みで塗布した。これを80℃で3時間硬化し、積層体を得た。得られた積層体の塗膜は、衝撃に強い塗膜であった。更に、この積層体の塗膜を室外の日当たりのよい場所に1年間放置したが、塗膜の黄変は観察されなかった。
実施例1の硬化性樹脂組成物を用いて、特表2002-530445号公報の実施例に従って、ガラス繊維強化複合材料を作製した。得られた複合材料は、高い靱性を示した。
Claims (37)
- 平均水酸基価が200~1500mgKOH/gであるポリオール(A)と、ポリイソシアネート(B)と、ポリマー微粒子(C)を含有することを特徴とする硬化性樹脂組成物。
- 前記ポリオール(A)として、ポリエーテルポリオール(a1)を含有することを特徴とする請求項1に記載の硬化性樹脂組成物。
- 前記ポリオール(A)の平均水酸基価が350~1500mgKOH/gであり、さらに硬化触媒(D)を含有することを特徴とする請求項1または2に記載の硬化性樹脂組成物。
- 前記ポリエーテルポリオール(a1)の量が、ポリオール(A)成分の総量100質量部の内、50質量部以上であることを特徴とする請求項2または3に記載の硬化性樹脂組成物。
- ポリオール(A)と、ポリイソシアネート(B)と、ポリマー微粒子(C)を含有し、前記ポリオール(A)として、ポリエステルポリオール(a2)を必ず含有し、かつポリエステルポリオール(a2)の量が、ポリオール(A)成分の総量100質量部の内、20質量部以上であることを特徴とする硬化性樹脂組成物。
- 前記ポリオール(A)の平均水酸基価が20~1000mgKOH/gであることを特徴とする請求項5に記載の硬化性樹脂組成物。
- さらに硬化触媒(D)を含有する請求項5または6に記載の硬化性樹脂組成物。
- 前記ポリイソシアネート(B)を、前記ポリオール(A)100質量部に対して、2~5000質量部含有し、
前記ポリマー微粒子(C)を、前記ポリオール(A)と前記ポリイソシアネート(B)の総量100質量部に対して、1~100質量部含有することを特徴とする請求項1~7のいずれかに記載の硬化性樹脂組成物。 - 前記(C)成分の体積平均粒子径が、10~2000nmであることを特徴とする請求項1~8のいずれかに記載の硬化性樹脂組成物。
- 前記(C)成分が、コアシェル構造を有することを特徴とする請求項1~9のいずれかに記載の硬化性樹脂組成物。
- 前記(C)成分が、ジエン系ゴム、(メタ)アクリレート系ゴム、及びオルガノシロキサン系ゴムよりなる群から選択される1種以上のコア層を有することを特徴とする請求項1~10のいずれかに記載の硬化性樹脂組成物。
- 前記ジエン系ゴムが、ブタジエンゴム、およびブタジエン-スチレンゴムよりなる群から選択される1種以上であることを特徴とする請求項11に記載の硬化性樹脂組成物。
- 前記(C)成分が、芳香族ビニルモノマー、ビニルシアンモノマー、及び(メタ)アクリレートモノマーよりなる群から選択される1種以上のモノマー成分を、コア層にグラフト重合してなるシェル層を有することを特徴とする請求項1~12のいずれかに記載の硬化性樹脂組成物。
- 前記(C)成分が、水酸基を有するモノマー成分を、コア層にグラフト重合してなるシェル層を有することを特徴とする請求項1~13のいずれかに記載の硬化性樹脂組成物。
- 前記(C)成分が、該硬化性樹脂組成物中で1次粒子の状態で分散していることを特徴とする請求項1~14のいずれかに記載の硬化性樹脂組成物。
- 前記(A)成分が、3官能以上の多官能ポリオールを含有することを特徴とする請求項1~15のいずれかに記載の硬化性樹脂組成物。
- 前記(A)成分の総量100質量部の内、3官能以上の多官能ポリオールの含有量が20質量部以上であることを特徴とする請求項16に記載の硬化性樹脂組成物。
- 前記(B)成分が、環状構造または直鎖構造もしくは分岐鎖構造を有するポリイソシアネートであることを特徴とする請求項1~17のいずれかに記載の硬化性樹脂組成物。
- 前記(B)成分が、芳香族ポリイソシアネートであることを特徴とする請求項18に記載の硬化性樹脂組成物。
- 前記(B)成分が、イソシアネート基を1分子当り平均して2.1個以上有するポリイソシアネートであることを特徴とする請求項1~19のいずれかに記載の硬化性樹脂組成物。
- 前記(B)成分が、脂環族ポリイソシアネート、および脂肪族ポリイソシアネートよりなる群から選択される1種以上であることを特徴とする請求項18に記載の硬化性樹脂組成物。
- 前記(B)成分が、脂環族ポリイソシアネートである請求項21に記載の硬化性樹脂組成物。
- 前記(A)成分と前記(B)成分の総量100質量部に対して、硬化触媒(D)0.001~20質量部をさらに含有することを特徴とする請求項1~22のいずれかに記載の硬化性樹脂組成物。
- 前記(D)成分が有機スズ化合物であることを特徴とする請求項23に記載の硬化性樹脂組成物。
- 前記(A)成分と前記(B)成分の総量100質量部に対して、脱水剤(E)0.1~30質量部をさらに含有することを特徴とする請求項1~24のいずれかに記載の硬化性樹脂組成物。
- 前記(B)成分のイソシアネート基の総モル量(β)と前記(A)成分の水酸基の総モル量(α)との比の値(β/α)が、0.7~1.5であることを特徴とする請求項1~25のいずれかに記載の硬化性樹脂組成物。
- 前記(A)成分と前記(B)成分との反応により得られるウレタンプレポリマーを含有することを特徴とする請求項1~26のいずれかに記載の硬化性樹脂組成物。
- 前記(B)成分のイソシアネート(NCO)基と前記(A)成分の活性水素含有基との当量比(NCO/活性水素含有基)が、1.05~5.0の範囲で反応して得たイソシアネートを有するウレタンプレポリマーを含有することを特徴とする請求項27に記載の硬化性樹脂組成物。
- 請求項28に記載の硬化性樹脂組成物からなる1液型湿分硬化性樹脂組成物。
- 請求項1~28のいずれかに記載の硬化性樹脂組成物であって、前記(B)成分を含有する第1液と、前記(A)成分と前記(C)成分と前記(D)成分を含有する第2液とからなる2液型硬化性樹脂組成物。
- 請求項1~30のいずれかに記載の硬化性樹脂組成物を硬化して得られる硬化物。
- 請求項1~30のいずれかに記載の硬化性樹脂組成物を用いてなる構造接着剤。
- 請求項1~30のいずれかに記載の硬化性樹脂組成物を用いてなるコーティング材。
- 請求項1~30のいずれかに記載の硬化性樹脂組成物を、金属または多孔質下地に塗布した後、該硬化性樹脂組成物を硬化してなる積層体。
- 請求項1~30のいずれかに記載の硬化性樹脂組成物を、強化繊維のバインダーとして用いてなる繊維強化複合材料。
- 請求項1~30のいずれかに記載の硬化性樹脂組成物を発泡してなる発泡体。
- ガラス転移温度が75℃以上である請求項31に記載の硬化物。
Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2015521491A JP6463674B2 (ja) | 2013-06-07 | 2014-06-05 | 硬化性樹脂組成物、それを用いてなる構造接着剤、コーティング材又は繊維強化複合材料、それを発泡してなる発泡体、それを硬化してなる積層体、及びそれらの硬化物 |
| EP19190207.1A EP3604371B1 (en) | 2013-06-07 | 2014-06-05 | Curable resin composition, structural adhesive, coating material or fiber reinforced composite material using the same, foam body using the same, laminate using the same, and cured material thereof |
| US14/896,295 US10100195B2 (en) | 2013-06-07 | 2014-06-05 | Curable resin composition, structural adhesive, coating material or fiber reinforced composite material using the same, foam body using the same, laminate using the same, and cured material thereof |
| EP14806856.2A EP3006478B1 (en) | 2013-06-07 | 2014-06-05 | Curable resin composition, structural adhesive produced using same, coating material or fiber reinforced composite material, foam body produced by foaming same, laminated body produced by curing same, and cured product thereof |
| CN201480032569.4A CN105308085B (zh) | 2013-06-07 | 2014-06-05 | 固化性树脂组合物、使用其而成的结构粘接剂、涂覆材料或纤维强化复合材料、将其发泡而成的发泡体、将其固化而成的层叠体及它们的固化物 |
| US16/117,866 US10669419B2 (en) | 2013-06-07 | 2018-08-30 | Curable resin composition, structural adhesive, coating material or fiber reinforced composite material using the same, foam body using the same, laminate using the same, and cured material thereof |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2013-120388 | 2013-06-07 | ||
| JP2013120388 | 2013-06-07 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US14/896,295 A-371-Of-International US10100195B2 (en) | 2013-06-07 | 2014-06-05 | Curable resin composition, structural adhesive, coating material or fiber reinforced composite material using the same, foam body using the same, laminate using the same, and cured material thereof |
| US16/117,866 Division US10669419B2 (en) | 2013-06-07 | 2018-08-30 | Curable resin composition, structural adhesive, coating material or fiber reinforced composite material using the same, foam body using the same, laminate using the same, and cured material thereof |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2014196607A1 true WO2014196607A1 (ja) | 2014-12-11 |
Family
ID=52008238
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2014/064998 WO2014196607A1 (ja) | 2013-06-07 | 2014-06-05 | 硬化性樹脂組成物、それを用いてなる構造接着剤、コーティング材又は繊維強化複合材料、それを発泡してなる発泡体、それを硬化してなる積層体、及びそれらの硬化物 |
Country Status (5)
| Country | Link |
|---|---|
| US (2) | US10100195B2 (ja) |
| EP (2) | EP3604371B1 (ja) |
| JP (1) | JP6463674B2 (ja) |
| CN (1) | CN105308085B (ja) |
| WO (1) | WO2014196607A1 (ja) |
Cited By (26)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20150204476A1 (en) * | 2014-01-23 | 2015-07-23 | Neptune Research, Inc. | Unidirectional fiber composite system for structural repairs and reinforcement |
| WO2015156295A1 (ja) * | 2014-04-07 | 2015-10-15 | 株式会社カネカ | 熱可塑性ポリウレタン系樹脂組成物、導体被覆材及びこれらの製造方法 |
| JP2016113588A (ja) * | 2014-12-17 | 2016-06-23 | ヘンケルジャパン株式会社 | 積層シート用接着剤 |
| US20160214781A1 (en) * | 2015-01-22 | 2016-07-28 | Neptune Research, Inc. | Composite reinforcement systems and methods of manufacturing the same |
| JP2017082118A (ja) * | 2015-10-29 | 2017-05-18 | 横浜ゴム株式会社 | 2液ウレタン系接着剤組成物 |
| WO2017099196A1 (ja) * | 2015-12-11 | 2017-06-15 | 株式会社カネカ | 機械的強度に優れるポリマー微粒子含有ポリウレタン系硬化性組成物 |
| JP2017137387A (ja) * | 2016-02-02 | 2017-08-10 | 凸版印刷株式会社 | 非吸着性接着コーティング剤、非吸着性積層体、及び包装袋 |
| WO2017145953A1 (ja) * | 2016-02-22 | 2017-08-31 | 株式会社カネカ | ポリオール組成物および熱硬化性樹脂 |
| JP2017197696A (ja) * | 2016-04-28 | 2017-11-02 | 株式会社イノアックコーポレーション | 樹脂接着剤 |
| JP2018027993A (ja) * | 2016-08-15 | 2018-02-22 | 株式会社カネカ | 耐チッピング性に優れるポリマー微粒子含有塗料組成物 |
| JP2018039900A (ja) * | 2016-09-07 | 2018-03-15 | 株式会社カネカ | コアシェル構造を有するポリマー微粒子含有軟質ポリウレタンフォーム用硬化性組成物 |
| JP2018076662A (ja) * | 2016-11-07 | 2018-05-17 | 中国塗料株式会社 | 変位絶縁材組成物、硬化体、制水設備の変位絶縁方法および制水設備 |
| JP2018516782A (ja) * | 2015-05-11 | 2018-06-28 | コベストロ、リミテッド、ライアビリティ、カンパニーCovestro Llc | ポリウレタン樹脂を用いたフィラメントワインディング法および組成物を製造するためのシステム |
| US10017673B2 (en) | 2015-04-09 | 2018-07-10 | Kaneka Corporation | Polymer fine particle-containing curable resin composition having improved bonding strength against impact peeling |
| WO2019049864A1 (ja) * | 2017-09-08 | 2019-03-14 | オリジン電気株式会社 | ポリウレタン塗料組成物及び塗装製品の製造方法 |
| WO2019208569A1 (ja) | 2018-04-27 | 2019-10-31 | 株式会社カネカ | 作業性に優れるポリマー微粒子含有硬化性樹脂組成物を用いる接着方法、及び、該接着方法を用いて得られる積層体 |
| KR20190130704A (ko) * | 2018-05-14 | 2019-11-25 | 송금용 | 밀폐성 및 난연성이 향상된 코팅액 및 이를 코팅한 액체 보관 용기 |
| US10669419B2 (en) | 2013-06-07 | 2020-06-02 | Kaneka Corporation | Curable resin composition, structural adhesive, coating material or fiber reinforced composite material using the same, foam body using the same, laminate using the same, and cured material thereof |
| KR20200069742A (ko) * | 2018-12-07 | 2020-06-17 | 주식회사 엘지화학 | 경화성 조성물 |
| JP2021513591A (ja) * | 2018-02-07 | 2021-05-27 | ビーエイエスエフ・ソシエタス・エウロパエアBasf Se | 熱絶縁材料、接着剤および外層の複合要素 |
| JP2021531393A (ja) * | 2018-07-25 | 2021-11-18 | エルジー・ケム・リミテッド | 接着剤組成物 |
| WO2022191143A1 (ja) * | 2021-03-08 | 2022-09-15 | ハリマ化成株式会社 | 二液硬化型コーティング剤及び多層膜 |
| WO2023008175A1 (ja) * | 2021-07-26 | 2023-02-02 | 横浜ゴム株式会社 | 2液硬化型接着剤組成物 |
| WO2023038011A1 (ja) * | 2021-09-10 | 2023-03-16 | 積水化学工業株式会社 | 硬化性樹脂材料、樹脂成形体及び樹脂成形体の製造方法 |
| JP7379043B2 (ja) | 2019-09-24 | 2023-11-14 | 大日本塗料株式会社 | 2液型ウレタン塗料組成物 |
| JP7440241B2 (ja) | 2019-10-28 | 2024-02-28 | 株式会社イノアックコーポレーション | ポリウレタンフォーム |
Families Citing this family (32)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB2514139A (en) | 2013-05-14 | 2014-11-19 | Aghababaie Lin & Co Ltd | Apparatus for fabrication of three dimensional objects |
| US9976027B2 (en) * | 2013-10-29 | 2018-05-22 | Kaneka Corporation | Polymer fine particle-containing curable resin composition having improved storage stability |
| US10166725B2 (en) | 2014-09-08 | 2019-01-01 | Holo, Inc. | Three dimensional printing adhesion reduction using photoinhibition |
| JP6808491B2 (ja) * | 2014-10-21 | 2021-01-06 | 株式会社カネカ | 硬化性樹脂組成物及び硬化物 |
| US10651382B2 (en) * | 2015-03-30 | 2020-05-12 | Merck Patent Gmbh | Formulation of an organic functional material comprising a siloxane solvent |
| US11141919B2 (en) | 2015-12-09 | 2021-10-12 | Holo, Inc. | Multi-material stereolithographic three dimensional printing |
| CN107915821B (zh) * | 2016-10-10 | 2021-01-15 | 万华化学(北京)有限公司 | 一种聚氨酯泡沫及其制备方法和用途 |
| CN106391992B (zh) * | 2016-10-12 | 2018-07-13 | 山东科技大学 | 聚酯多元醇与酚醛树脂复合型冷芯盒制芯粘结剂体系 |
| JP6867793B2 (ja) * | 2016-12-14 | 2021-05-12 | 株式会社きもと | 遮光性摺動フィルム、遮光性摺動部材、及び遮光性摺動フィルム用樹脂組成物 |
| US10935891B2 (en) | 2017-03-13 | 2021-03-02 | Holo, Inc. | Multi wavelength stereolithography hardware configurations |
| GB2564956B (en) | 2017-05-15 | 2020-04-29 | Holo Inc | Viscous film three-dimensional printing systems and methods |
| US10245785B2 (en) | 2017-06-16 | 2019-04-02 | Holo, Inc. | Methods for stereolithography three-dimensional printing |
| EP3428216B1 (en) * | 2017-07-11 | 2022-11-02 | Henkel AG & Co. KGaA | Method for producing functionalized polyesters |
| CN107502235A (zh) * | 2017-08-18 | 2017-12-22 | 佛山市永恒达新材料科技有限公司 | 一种防水涂泥用粘合剂的制备方法 |
| SG11202003894SA (en) * | 2017-11-03 | 2020-05-28 | Gcp Applied Tech Inc | Pre-applied membrane having granular polymer outer protective layer |
| CN108610996A (zh) * | 2018-03-29 | 2018-10-02 | 重庆渝茁科技有限公司 | 轻质隔音型塑料蜂窝板材料 |
| KR20210022586A (ko) * | 2018-06-25 | 2021-03-03 | 디아이씨 가부시끼가이샤 | 수성 수지 조성물 및 그 제조 방법 |
| JP6516056B1 (ja) * | 2018-08-29 | 2019-05-22 | 東洋インキScホールディングス株式会社 | ウレタン系粘着剤および粘着シート |
| CN113474147A (zh) | 2018-12-26 | 2021-10-01 | 霍洛公司 | 用于三维打印系统和方法的传感器 |
| CN109762387B (zh) * | 2019-01-29 | 2021-05-18 | 佛山市顺德区龙江镇万安涂料有限公司 | 一种涂料组合物及其制备工艺 |
| RU2719959C1 (ru) * | 2019-06-13 | 2020-04-23 | Ао "ивхимпром" | Способ получения полимерной композиции (варианты) |
| WO2021133585A1 (en) * | 2019-12-23 | 2021-07-01 | Carbon, Inc. | Inhibition of crystallization in polyurethane resins |
| WO2022200180A1 (en) | 2021-03-24 | 2022-09-29 | Covestro Deutschland Ag | Method for preparing a polyurethane composite material by vacuum infusion process |
| EP4083101A1 (en) | 2021-04-29 | 2022-11-02 | Covestro Deutschland AG | Method for preparing a polyurethane composite material by vacuum infusion process |
| CN113593817B (zh) * | 2021-08-03 | 2022-07-22 | 包头天和磁材科技股份有限公司 | 磁体预制件、磁体组件及其制备方法 |
| CN114134751B (zh) * | 2021-11-01 | 2023-06-27 | 山东亿森美居新材料科技有限公司 | 一种强化木地板表层浸渍用耐水复合树脂及其制备方法 |
| CN114276070B (zh) * | 2021-12-30 | 2023-04-25 | 嘉兴正合科技有限公司 | 结构修复用超强高韧性聚酯混凝土、制备方法和应用 |
| CN114349402B (zh) * | 2021-12-30 | 2023-01-20 | 北京中科嘉固科技有限公司 | 结构修复用超强高韧性聚酯混凝土、制备方法和应用 |
| CN114149200B (zh) * | 2021-12-30 | 2022-11-22 | 嘉兴正合科技有限公司 | 结构快速修复用超强高韧性聚酯混凝土、制备方法和应用 |
| WO2023247584A1 (en) | 2022-06-24 | 2023-12-28 | Zephyros, Inc. | Thermal runaway fumes management |
| KR20240018238A (ko) * | 2022-08-02 | 2024-02-13 | 주식회사 엘지화학 | 광변조 디바이스 |
| CN115215987B (zh) * | 2022-08-22 | 2024-01-23 | 安徽大学 | 高固含量纳米碳酸钙复合聚氨酯泡沫及其制备方法 |
Citations (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS55157620A (en) | 1979-05-25 | 1980-12-08 | Bayer Ag | Thermosetting forming composition and manufacture of product |
| JPH0349916A (ja) | 1989-07-19 | 1991-03-04 | Fuji Xerox Co Ltd | 複合材の成形方法及びこれに用いる複合同時成形金型並びに複合材 |
| WO2000029459A1 (en) | 1998-11-16 | 2000-05-25 | Huntsman International Llc | Polyisocyanurate compositions and composites |
| JP2003155389A (ja) | 2001-11-22 | 2003-05-27 | Sunstar Eng Inc | 加水分解性シリル基含有硬化性組成物 |
| US20030134085A1 (en) | 2001-12-17 | 2003-07-17 | Peter Haas | Laminated parts made of outer layers and polyurethane sandwich materials and their production |
| WO2005028546A1 (ja) | 2003-09-18 | 2005-03-31 | Kaneka Corporation | ゴム状重合体粒子の製造方法およびこれを含有する樹脂組成物の製造方法 |
| WO2006070664A1 (ja) | 2004-12-28 | 2006-07-06 | Kaneka Corporation | グラフト共重合体およびその製造方法、並びに該グラフト共重合体含有樹脂組成物 |
| US20060173128A1 (en) | 2004-11-15 | 2006-08-03 | Huntsman International Llc | Pultrusion systems and process |
| US20070098997A1 (en) | 2005-11-02 | 2007-05-03 | Bayer Materialscience Llc | Composite articles and a process for their production |
| WO2009014037A1 (ja) | 2007-07-25 | 2009-01-29 | Kaneka Corporation | 樹脂組成物、および該樹脂組成物を用いた重縮合体 |
| JP2010116429A (ja) * | 2008-11-11 | 2010-05-27 | Sanyo Chem Ind Ltd | ウレタン樹脂用ポリマーポリオール組成物 |
| WO2011028271A2 (en) | 2009-09-04 | 2011-03-10 | Bayer Materialscience Llc | Automated processes for the production of polyurethane wind turbine blades |
| JP2011190286A (ja) * | 2010-03-11 | 2011-09-29 | Yokohama Rubber Co Ltd:The | ホットメルト接着剤組成物 |
| US20120245286A1 (en) | 2011-03-25 | 2012-09-27 | Bayer Materialscience Llc | Polyurethane composites produced by a vacuum infusion process |
| JP2012251053A (ja) | 2011-06-01 | 2012-12-20 | Yokohama Rubber Co Ltd:The | ウレタン樹脂接着剤組成物 |
Family Cites Families (27)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB2244714B (en) * | 1990-05-31 | 1993-10-06 | Sanyo Chemical Ind Ltd | Foamed polyurethane-forming composition,foamed polyurethane and process for making the same |
| JPH11349916A (ja) | 1998-06-05 | 1999-12-21 | Sunstar Eng Inc | 高揺変性変成シリコーン系接着剤 |
| US6159405A (en) * | 1998-09-22 | 2000-12-12 | Borden Chemical, Inc. | Phenolic resin system for pultrusion composites |
| US7202302B2 (en) | 1998-11-16 | 2007-04-10 | Huntsman International Llc | Polyisocyanurate compositions and composites |
| DE10122437A1 (de) * | 2001-05-09 | 2002-11-28 | Henkel Kgaa | Schmelzklebstoff in Form eines Granulates |
| US7087672B2 (en) * | 2002-05-08 | 2006-08-08 | E. I. Du Pont De Nemours And Company | Non-yellowing polyester coating composition |
| EP1728832A4 (en) * | 2004-03-23 | 2013-06-05 | Sanyo Chemical Ind Ltd | RESIN BODIES FOR CUTTING OPERATION, MATERIAL FOR ITS MANUFACTURE AND MODEL |
| JP2005272647A (ja) | 2004-03-25 | 2005-10-06 | Aisin Chem Co Ltd | 構造用接着剤組成物 |
| JP4639766B2 (ja) | 2004-11-16 | 2011-02-23 | 横浜ゴム株式会社 | 二液型常温硬化性エポキシ樹脂組成物および金属接着剤組成物 |
| EP1690880A1 (de) | 2005-02-11 | 2006-08-16 | Sika Technology AG | Zweikomponentige Polyurethanzusammensetzungen, insbesondere geeignet für den Einsatz als strukturelle Klebstoffe |
| PT2066717E (pt) | 2006-09-15 | 2009-12-21 | Basf Se | Processo para a preparação de espumas rígidas de poliuretano |
| EP1916269A1 (de) * | 2006-10-24 | 2008-04-30 | Sika Technology AG | Blockierte Polyurethanprepolymere und hitzehärtende Epoxidharzzusammensetzungen |
| ATE434653T1 (de) * | 2006-11-17 | 2009-07-15 | Sika Technology Ag | Polyaldimin enthaltende feuchtigkeitshärtende heissschmelzklebstoff-zusammensetzung |
| EP2085414A4 (en) * | 2006-11-20 | 2012-11-07 | Asahi Glass Co Ltd | PROCESS FOR PRODUCING HARD POLYURETHANE FOAM |
| EP1975187A1 (de) * | 2007-03-28 | 2008-10-01 | Sika Technology AG | Verfahren zur Herstellung von Polyurethanzusammensetzungen mit niedrigem Isocyanat-Monomergehalt |
| EP2154168A4 (en) * | 2007-06-07 | 2011-12-28 | Asahi Glass Co Ltd | RESIN COMPOSITION CONTAINING THERMOPLASTIC POLYURETHANE AND THERMOFUSIBLE ADHESIVE |
| JP4851503B2 (ja) * | 2007-10-10 | 2012-01-11 | 三洋化成工業株式会社 | 微粒子分散ポリオールの製造方法及びポリウレタン樹脂の製造方法 |
| JP5086774B2 (ja) | 2007-11-01 | 2012-11-28 | サンスター技研株式会社 | 構造用接着剤 |
| JP5459206B2 (ja) * | 2008-05-30 | 2014-04-02 | 旭硝子株式会社 | 硬質発泡合成樹脂およびその製造方法 |
| JP5412952B2 (ja) * | 2009-05-20 | 2014-02-12 | 横浜ゴム株式会社 | エポキシ樹脂組成物 |
| JP5609877B2 (ja) * | 2009-08-27 | 2014-10-22 | 旭硝子株式会社 | ホットメルト接着剤組成物 |
| EP2650318A4 (en) | 2010-12-07 | 2014-09-24 | Asahi Glass Co Ltd | PROCESS FOR PRODUCING POLYETHER POLYOL AND PROCESS FOR PRODUCING RIGID FOAM SYNTHESIS RESIN |
| GB201102672D0 (en) * | 2011-02-15 | 2011-03-30 | Zephyros Inc | Improved structural adhesives |
| DE102011056166A1 (de) | 2011-12-08 | 2013-06-13 | Universität Stuttgart | Elektrooptischer Phasenmodulator |
| EP2871197B1 (en) * | 2012-07-05 | 2017-01-25 | Hitachi Chemical Co., Ltd. | Phenolic resin composition |
| JP2014083904A (ja) | 2012-10-22 | 2014-05-12 | Daikyonishikawa Corp | 車両用外装パネル |
| US10100195B2 (en) | 2013-06-07 | 2018-10-16 | Kaneka Corporation | Curable resin composition, structural adhesive, coating material or fiber reinforced composite material using the same, foam body using the same, laminate using the same, and cured material thereof |
-
2014
- 2014-06-05 US US14/896,295 patent/US10100195B2/en active Active
- 2014-06-05 CN CN201480032569.4A patent/CN105308085B/zh active Active
- 2014-06-05 EP EP19190207.1A patent/EP3604371B1/en active Active
- 2014-06-05 JP JP2015521491A patent/JP6463674B2/ja active Active
- 2014-06-05 WO PCT/JP2014/064998 patent/WO2014196607A1/ja active Application Filing
- 2014-06-05 EP EP14806856.2A patent/EP3006478B1/en active Active
-
2018
- 2018-08-30 US US16/117,866 patent/US10669419B2/en active Active
Patent Citations (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS55157620A (en) | 1979-05-25 | 1980-12-08 | Bayer Ag | Thermosetting forming composition and manufacture of product |
| US4251428A (en) | 1979-05-25 | 1981-02-17 | Bayer Aktiengesellschaft | Thermosetting molding compositions containing polyurethane and a fibrous material, and a process for the production of moldings |
| JPH0349916A (ja) | 1989-07-19 | 1991-03-04 | Fuji Xerox Co Ltd | 複合材の成形方法及びこれに用いる複合同時成形金型並びに複合材 |
| WO2000029459A1 (en) | 1998-11-16 | 2000-05-25 | Huntsman International Llc | Polyisocyanurate compositions and composites |
| JP2002530445A (ja) | 1998-11-16 | 2002-09-17 | ハンツマン・インターナショナル・エルエルシー | ポリイソシアヌレート組成物及び複合材 |
| JP2003155389A (ja) | 2001-11-22 | 2003-05-27 | Sunstar Eng Inc | 加水分解性シリル基含有硬化性組成物 |
| US20030134085A1 (en) | 2001-12-17 | 2003-07-17 | Peter Haas | Laminated parts made of outer layers and polyurethane sandwich materials and their production |
| JP2003220661A (ja) | 2001-12-17 | 2003-08-05 | Bayer Ag | 積層品およびその製法 |
| WO2005028546A1 (ja) | 2003-09-18 | 2005-03-31 | Kaneka Corporation | ゴム状重合体粒子の製造方法およびこれを含有する樹脂組成物の製造方法 |
| US20060173128A1 (en) | 2004-11-15 | 2006-08-03 | Huntsman International Llc | Pultrusion systems and process |
| WO2006070664A1 (ja) | 2004-12-28 | 2006-07-06 | Kaneka Corporation | グラフト共重合体およびその製造方法、並びに該グラフト共重合体含有樹脂組成物 |
| US20070098997A1 (en) | 2005-11-02 | 2007-05-03 | Bayer Materialscience Llc | Composite articles and a process for their production |
| JP2007125889A (ja) | 2005-11-02 | 2007-05-24 | Bayer Material Science Llc | 複合品およびその製造法 |
| WO2009014037A1 (ja) | 2007-07-25 | 2009-01-29 | Kaneka Corporation | 樹脂組成物、および該樹脂組成物を用いた重縮合体 |
| JP2010116429A (ja) * | 2008-11-11 | 2010-05-27 | Sanyo Chem Ind Ltd | ウレタン樹脂用ポリマーポリオール組成物 |
| WO2011028271A2 (en) | 2009-09-04 | 2011-03-10 | Bayer Materialscience Llc | Automated processes for the production of polyurethane wind turbine blades |
| JP2013504007A (ja) | 2009-09-04 | 2013-02-04 | バイエル・マテリアルサイエンス・リミテッド・ライアビリティ・カンパニー | ポリウレタンタービン翼のための自動化された製造方法 |
| JP2011190286A (ja) * | 2010-03-11 | 2011-09-29 | Yokohama Rubber Co Ltd:The | ホットメルト接着剤組成物 |
| US20120245286A1 (en) | 2011-03-25 | 2012-09-27 | Bayer Materialscience Llc | Polyurethane composites produced by a vacuum infusion process |
| JP2012251053A (ja) | 2011-06-01 | 2012-12-20 | Yokohama Rubber Co Ltd:The | ウレタン樹脂接着剤組成物 |
Non-Patent Citations (2)
| Title |
|---|
| JOURNAL OF THE SOCIETY OF RUBBER INDUSTRY, JAPAN, vol. 68, 1995, pages 417 |
| See also references of EP3006478A4 |
Cited By (45)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10669419B2 (en) | 2013-06-07 | 2020-06-02 | Kaneka Corporation | Curable resin composition, structural adhesive, coating material or fiber reinforced composite material using the same, foam body using the same, laminate using the same, and cured material thereof |
| US10953625B2 (en) * | 2014-01-23 | 2021-03-23 | Spartan Acquisition Llc | Unidirectional fiber composite system for structural repairs and reinforcement |
| US20150204476A1 (en) * | 2014-01-23 | 2015-07-23 | Neptune Research, Inc. | Unidirectional fiber composite system for structural repairs and reinforcement |
| US10814584B2 (en) | 2014-01-23 | 2020-10-27 | Neptune Research, Llc | Kits and methods for fiber composites including partially-cured resinous materials for the reinforcement of physical structures |
| WO2015156295A1 (ja) * | 2014-04-07 | 2015-10-15 | 株式会社カネカ | 熱可塑性ポリウレタン系樹脂組成物、導体被覆材及びこれらの製造方法 |
| JPWO2015156295A1 (ja) * | 2014-04-07 | 2017-04-13 | 株式会社カネカ | 熱可塑性ポリウレタン系樹脂組成物、導体被覆材及びこれらの製造方法 |
| US10311995B2 (en) | 2014-04-07 | 2019-06-04 | Kaneka Corporation | Thermoplastic polyurethane resin composition, conductor covering material, and manufacturing method of these |
| JP2016113588A (ja) * | 2014-12-17 | 2016-06-23 | ヘンケルジャパン株式会社 | 積層シート用接着剤 |
| US20160214781A1 (en) * | 2015-01-22 | 2016-07-28 | Neptune Research, Inc. | Composite reinforcement systems and methods of manufacturing the same |
| US10597182B2 (en) | 2015-01-22 | 2020-03-24 | Neptune Research, Llc. | Composite reinforcement systems and methods of manufacturing the same |
| EP3247933A4 (en) * | 2015-01-22 | 2018-10-24 | Neptune Research, Llc | Composite reinforcement systems and methods of manufacturing the same |
| US11453518B2 (en) | 2015-01-22 | 2022-09-27 | Csc Operating Company, Llc | Composite reinforcement systems and methods of manufacturing the same |
| US10017673B2 (en) | 2015-04-09 | 2018-07-10 | Kaneka Corporation | Polymer fine particle-containing curable resin composition having improved bonding strength against impact peeling |
| JP2018516782A (ja) * | 2015-05-11 | 2018-06-28 | コベストロ、リミテッド、ライアビリティ、カンパニーCovestro Llc | ポリウレタン樹脂を用いたフィラメントワインディング法および組成物を製造するためのシステム |
| JP2017082118A (ja) * | 2015-10-29 | 2017-05-18 | 横浜ゴム株式会社 | 2液ウレタン系接着剤組成物 |
| CN108368335B (zh) * | 2015-12-11 | 2021-09-28 | 株式会社钟化 | 机械强度优异的含有聚合物微粒的聚氨酯系固化性组合物 |
| CN108368335A (zh) * | 2015-12-11 | 2018-08-03 | 株式会社钟化 | 机械强度优异的含有聚合物微粒的聚氨酯系固化性组合物 |
| JPWO2017099196A1 (ja) * | 2015-12-11 | 2018-11-01 | 株式会社カネカ | 機械的強度に優れるポリマー微粒子含有ポリウレタン系硬化性組成物 |
| US20180371238A1 (en) * | 2015-12-11 | 2018-12-27 | Kaneka Corporation | Polyurethane curable composition containing polymer fine particles excellent in mechanical strength |
| EP3388487A4 (en) * | 2015-12-11 | 2019-09-04 | Kaneka Corporation | HARDENABLE POLYURETHANE COMPOSITION WITH FINE POLYMER PARTICLES WITH EXCELLENT MECHANICAL STRENGTH |
| US10920073B2 (en) | 2015-12-11 | 2021-02-16 | Kaneka Corporation | Polyurethane curable composition containing polymer fine particles excellent in mechanical strength |
| WO2017099196A1 (ja) * | 2015-12-11 | 2017-06-15 | 株式会社カネカ | 機械的強度に優れるポリマー微粒子含有ポリウレタン系硬化性組成物 |
| JP2017137387A (ja) * | 2016-02-02 | 2017-08-10 | 凸版印刷株式会社 | 非吸着性接着コーティング剤、非吸着性積層体、及び包装袋 |
| CN108699329A (zh) * | 2016-02-22 | 2018-10-23 | 株式会社钟化 | 多元醇组合物和热固性树脂 |
| JPWO2017145953A1 (ja) * | 2016-02-22 | 2018-12-13 | 株式会社カネカ | ポリオール組成物および熱硬化性樹脂 |
| WO2017145953A1 (ja) * | 2016-02-22 | 2017-08-31 | 株式会社カネカ | ポリオール組成物および熱硬化性樹脂 |
| JP2017197696A (ja) * | 2016-04-28 | 2017-11-02 | 株式会社イノアックコーポレーション | 樹脂接着剤 |
| JP2018027993A (ja) * | 2016-08-15 | 2018-02-22 | 株式会社カネカ | 耐チッピング性に優れるポリマー微粒子含有塗料組成物 |
| JP2018039900A (ja) * | 2016-09-07 | 2018-03-15 | 株式会社カネカ | コアシェル構造を有するポリマー微粒子含有軟質ポリウレタンフォーム用硬化性組成物 |
| JP2018076662A (ja) * | 2016-11-07 | 2018-05-17 | 中国塗料株式会社 | 変位絶縁材組成物、硬化体、制水設備の変位絶縁方法および制水設備 |
| WO2019049864A1 (ja) * | 2017-09-08 | 2019-03-14 | オリジン電気株式会社 | ポリウレタン塗料組成物及び塗装製品の製造方法 |
| JP2021513591A (ja) * | 2018-02-07 | 2021-05-27 | ビーエイエスエフ・ソシエタス・エウロパエアBasf Se | 熱絶縁材料、接着剤および外層の複合要素 |
| JP7391856B2 (ja) | 2018-02-07 | 2023-12-05 | ビーエーエスエフ ソシエタス・ヨーロピア | 熱絶縁材料、接着剤および外層の複合要素 |
| WO2019208569A1 (ja) | 2018-04-27 | 2019-10-31 | 株式会社カネカ | 作業性に優れるポリマー微粒子含有硬化性樹脂組成物を用いる接着方法、及び、該接着方法を用いて得られる積層体 |
| KR102072720B1 (ko) | 2018-05-14 | 2020-02-04 | 주식회사 지디프라코 | 밀폐성 및 난연성이 향상된 코팅액 및 이를 코팅한 액체 보관 용기 |
| KR20190130704A (ko) * | 2018-05-14 | 2019-11-25 | 송금용 | 밀폐성 및 난연성이 향상된 코팅액 및 이를 코팅한 액체 보관 용기 |
| JP7154378B2 (ja) | 2018-07-25 | 2022-10-17 | エルジー・ケム・リミテッド | 接着剤組成物 |
| JP2021531393A (ja) * | 2018-07-25 | 2021-11-18 | エルジー・ケム・リミテッド | 接着剤組成物 |
| KR102557201B1 (ko) * | 2018-12-07 | 2023-07-19 | 주식회사 엘지화학 | 경화성 조성물 |
| KR20200069742A (ko) * | 2018-12-07 | 2020-06-17 | 주식회사 엘지화학 | 경화성 조성물 |
| JP7379043B2 (ja) | 2019-09-24 | 2023-11-14 | 大日本塗料株式会社 | 2液型ウレタン塗料組成物 |
| JP7440241B2 (ja) | 2019-10-28 | 2024-02-28 | 株式会社イノアックコーポレーション | ポリウレタンフォーム |
| WO2022191143A1 (ja) * | 2021-03-08 | 2022-09-15 | ハリマ化成株式会社 | 二液硬化型コーティング剤及び多層膜 |
| WO2023008175A1 (ja) * | 2021-07-26 | 2023-02-02 | 横浜ゴム株式会社 | 2液硬化型接着剤組成物 |
| WO2023038011A1 (ja) * | 2021-09-10 | 2023-03-16 | 積水化学工業株式会社 | 硬化性樹脂材料、樹脂成形体及び樹脂成形体の製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| JPWO2014196607A1 (ja) | 2017-02-23 |
| EP3006478A4 (en) | 2017-03-01 |
| US20160122539A1 (en) | 2016-05-05 |
| EP3006478B1 (en) | 2019-09-04 |
| US10669419B2 (en) | 2020-06-02 |
| CN105308085A (zh) | 2016-02-03 |
| JP6463674B2 (ja) | 2019-02-06 |
| EP3604371A1 (en) | 2020-02-05 |
| EP3006478A1 (en) | 2016-04-13 |
| EP3604371B1 (en) | 2021-01-13 |
| US20180371239A1 (en) | 2018-12-27 |
| CN105308085B (zh) | 2017-12-01 |
| US10100195B2 (en) | 2018-10-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6463674B2 (ja) | 硬化性樹脂組成物、それを用いてなる構造接着剤、コーティング材又は繊維強化複合材料、それを発泡してなる発泡体、それを硬化してなる積層体、及びそれらの硬化物 | |
| JP6936734B2 (ja) | 機械的強度に優れるポリマー微粒子含有ポリウレタン系硬化性組成物 | |
| CN107075246B (zh) | 改性聚天冬氨酸酯和固化性树脂组合物 | |
| JP2018090651A (ja) | 貯蔵安定性に優れる硬化性エポキシ樹脂組成物 | |
| JP7271514B2 (ja) | 作業性に優れるポリマー微粒子含有硬化性樹脂組成物を用いる接着方法、及び、該接着方法を用いて得られる積層体 | |
| WO2020138263A1 (ja) | 樹脂組成物およびその利用 | |
| JP6722477B2 (ja) | 剥離接着性および耐衝撃剥離接着性の改善されたポリマー微粒子含有硬化性樹脂組成物 | |
| JP7290446B2 (ja) | 硬化性樹脂組成物およびその利用 | |
| JP2016104834A (ja) | コアシェルポリマーにより改質された変性熱可塑性ウレタンエラストマー | |
| JP7442992B2 (ja) | 積層体および積層体の製造方法 | |
| WO2020218552A1 (ja) | 構造物の製造方法 | |
| WO2020196920A1 (ja) | 樹脂組成物の製造方法および樹脂組成物 | |
| JP7277241B2 (ja) | 作業性に優れるポリマー微粒子含有硬化性樹脂組成物を用いる接着方法、及び、該接着方法を用いて得られる積層体 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201480032569.4 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 14806856 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2015521491 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14896295 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2014806856 Country of ref document: EP |